Applied Physics
Vol.1 No.3(2011), Article ID:280,5 pages DOI:10.4236/app.2011.13014
Effect of Si Adsorption on Graphene Doped withN, Al and P
1College of Physics and Electronic Engineering, Xuzhou Normal University, Xuzhou
2College of Physics and Information Engineering, Henan Normal University, Xinxiang
3Physics Department, Tongji University, Shanghai
Email: xuqing_qiang@sina.com; shke@tongji.edu.cn
Received: Jun. 2nd, 2011; revised: Jul. 12th, 2011; accepted: Jul. 17th, 2011.
ABSTRACT:
First-principles calculations based on density functional theory were carried out to study the effects of nitrogen, aluminum and phosphorus doping on Si adsorption on grapheme. We found that N doping is the n-type doping of grapheme and increases the conductivity of the grapheme system. Both N and P doping graphene system are relatively stable while Al doping graphene system is not. The effect is stronger for Si adsorption position on graphene for different types of impurity. Al and P atom enhance the Si adsorption on graphene, the effect of N doping is far less than the other two dopant do. No magnetic moment was observed in Si adsorbed on Al doping graphene while net magnetism was brought in Si adsorbed on N and P doping graphene.
Keywords: Graphene; Dopant; Adsorption; First-Principles
N、Al、P替位掺杂对Si在石墨烯上吸附的影响
胡功臣1,李艳慧2,徐庆强1*,柯三黄3*
1徐州师范大学物理与电子工程学院,徐州
2河南师范大学物理与信息工程学院,新乡
3上海同济大学物理系,上海
Email: xuqing_qiang@sina.com; shke@tongji.edu.cn
摘 要:
采用基于密度泛函理论的第一性原理计算了N、Al和P掺杂对Si在石墨烯上吸附的影响。结果表明:N掺杂石墨烯为n型掺杂,提高了石墨烯体系的导电性;N、P原子掺杂比Al掺杂石墨烯容易;杂质类型对Si在石墨烯上的吸附位置有显著的影响;Al、P掺杂增强了Si在石墨烯上的吸附, N掺杂对Si在石墨烯上吸附的影响很小;Si吸附在N-graphene、P-graphene体系具有磁性,Si吸附Al-graphene体系磁矩为零,不显示磁性。
收稿日期:2011年6月2日;修回日期:2011年7月12日;录用日期:2011年7月17日
关键词:石墨烯;掺杂;吸附;第一性原理
1. 引言
石墨烯具有规整的二维结构和良好的导电能力,自2004年被安德烈·K·海姆(Andre K. Geim)等人[1]制备出来就立刻引起了人们的广泛关注。石墨烯是由单层碳原子紧密堆积成二维蜂窝晶体结构的一种碳质新材料,可被看作富勒烯和碳纳米管的基础结构[2]。
对石墨烯的研究,最初主要集中在其制备和基本电子性质等方面[3,4],随后,研究人员又对碱金属、贵金属、过渡金属等在石墨烯上的吸附[5-7]以及非金属在石墨烯上的吸附[8,9]进行了一系列的深入研究。近年来,研究主要是通过不同杂质替代碳原子来观察石墨烯的结构性质,并实现石墨烯功能化,这些杂质的原子包括氮原子,硼原子,过渡金属元素(如铁,钴,镍,铑,铱)以及碱金属(锂和钾)[10-12]。原子掺杂会在能带结构中引入杂质能级,不同掺杂原子和掺杂方式对能级结构影响又不同。由于掺杂原子的存在,使碳材料的结构和性能发生了改变[13,14],杂质的掺杂调整了碳纳米结构的特性和化学敏感度,也为未来纳米微电子器件的选择制备提供了依据。
在元素周期表中,B和N位于C的两侧,具有相似的电子结构,而Si在两者分别掺杂的石墨烯上吸附后,除了Si稳定吸附位置不同外,Si在掺杂石墨烯上的形成能以及体系产生的磁矩也都有较大的差别。如,Si在B掺杂石墨烯上稳定吸附位置在距B原子正上方1.965 Å处[15],本文计算得出Si在N掺杂石墨烯上吸附位置在C-N键的中点上方2.181 Å处;B掺杂后Si在石墨烯上的形成能为3.053 eV[15],比Si吸附在N掺杂石墨烯上的形成能大2.016 eV;B掺杂石墨烯吸附Si后体系没有磁性[15],而N掺杂石墨烯吸附后体系产生磁矩为1.0 。此外,本文还对B同主族的Al、N同主族的P掺杂石墨烯以及Si在掺杂石墨烯上吸附后体系的电子性质和磁性变化方面做了研究。
。此外,本文还对B同主族的Al、N同主族的P掺杂石墨烯以及Si在掺杂石墨烯上吸附后体系的电子性质和磁性变化方面做了研究。
2. 计算模型和方法
本文选取4 × 4的石墨烯超胞,共32个原子,计算采用基于密度泛函理论(DFT)平面赝势方法的VASP程序包[16]完成。通常,采用局域密度近似(LDA)方法计算得到的结果键能会偏大,键长、键角则会偏小。因此,电子交换关联能函数我们采用广义梯度近似(GGA)中的PBE泛函形式[17],并选用超软赝势来描述离子实与价电子之间的相互作用。在倒易的k空间中,平面波截止能选取400 eV。计算中取X和Y方向在石墨烯平面内,Z方向垂直于石墨烯平面。真空层取1.5 nm。自洽迭代过程简约布里渊区积分k点选取Gamma 9 × 9 × 1 格点,计算中考虑了自旋极化效应,体系能量优化中总能量误差不大于0.001 eV。在石墨烯上我们主要取三个高对称吸附位置(如图1):心位(H)位于六方蜂巢格子的正中心;桥位(B)位于C-C键的中点;顶位(T)位于碳原子的正上方。
Si在石墨烯上吸附的平均形成能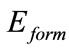 为:
为:

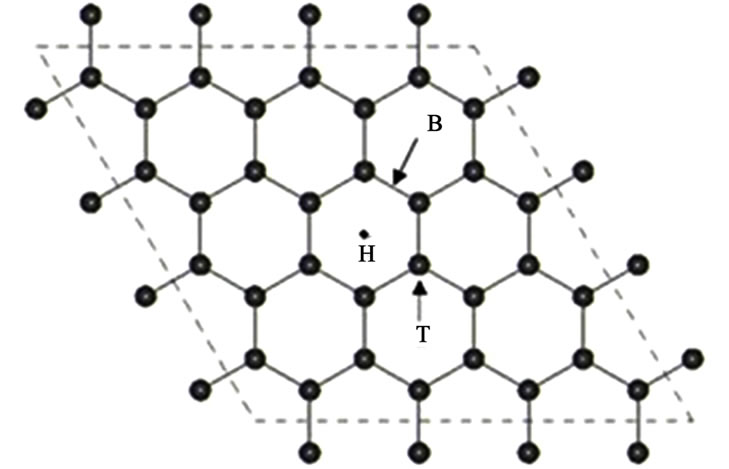
Figure 1. Schematic illustration for the three high symmetry sites on 4 × 4 cell of graphene surface. H: the hollow site, B: the bridge site, T: the top site
图1. 石墨烯4 × 4原胞的三个高对称吸附位置H(心位),B(桥位),T(顶位)
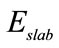 表示Si吸附前石墨烯衬底的总能量,
表示Si吸附前石墨烯衬底的总能量,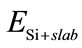 表示Si吸附后体系的总能,
表示Si吸附后体系的总能, 表示Si原子的能量,n为所吸附Si原子的个数。
表示Si原子的能量,n为所吸附Si原子的个数。
3. 结果和讨论
3.1. 替位掺杂石墨烯的结构和电子性质
通过替代石墨烯中一个碳原子来实现杂质的掺杂,氮(N)原子掺杂石墨烯记为N-graphene,铝(Al)原子掺杂石墨烯记为Al-graphene,磷(P)原子掺杂石墨烯记为P-graphene。N掺杂石墨烯与B掺杂石墨烯相似[18],N原子与衬底的C原子也在同一平面内,N原子替代C原子与周围C原子形成N-C键,与最近邻C原子距离dN-C为1.407 Å[18]。杂质原子Al、P为第三周期元素,原子半径比C原子大,不易与C原子形成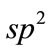 杂化,替位掺杂后引起石墨烯表面形成凸起,杂质周围C原子也发生移动,Al、P原子到最近邻碳原子的距离分别为dAl-C = 1.806 Ǻ,dP-C = 1.729 Ǻ。比较N、Al、P与C的键长,其中Al-C键长最长,N-C键长最短,这种结构的变化对石墨烯的活性产生一定影响。比较三种杂质掺杂后体系的形成能,Al不同于其它掺杂,Al-graphene体系具有较大的能量损失,其形成能最大[19],P掺杂形成能次之,N掺杂形成能最小。因此从掺杂结构和形成能的角度,N、P原子化学掺杂石墨烯比Al掺杂石墨烯容易。
杂化,替位掺杂后引起石墨烯表面形成凸起,杂质周围C原子也发生移动,Al、P原子到最近邻碳原子的距离分别为dAl-C = 1.806 Ǻ,dP-C = 1.729 Ǻ。比较N、Al、P与C的键长,其中Al-C键长最长,N-C键长最短,这种结构的变化对石墨烯的活性产生一定影响。比较三种杂质掺杂后体系的形成能,Al不同于其它掺杂,Al-graphene体系具有较大的能量损失,其形成能最大[19],P掺杂形成能次之,N掺杂形成能最小。因此从掺杂结构和形成能的角度,N、P原子化学掺杂石墨烯比Al掺杂石墨烯容易。
如图2所示为完整与掺杂石墨烯体系的态密度(DOS)图,与完整石墨烯相比,由于N比C多一个电
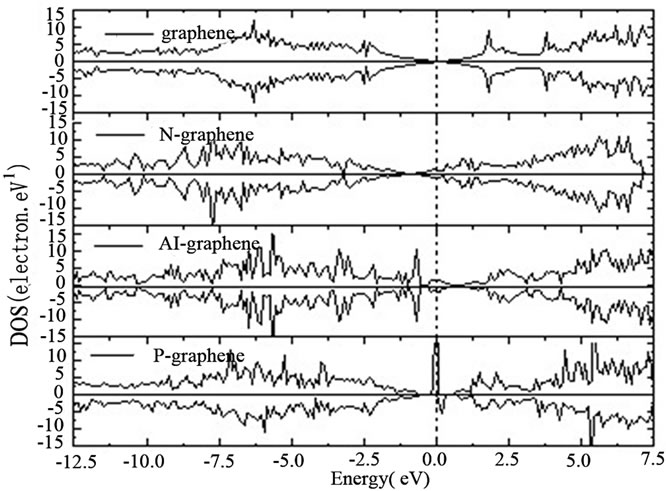
Figure 2. Density of states for perfect and doped graphene system
图2. 完整与掺杂石墨烯体系的态密度(DOS)图
子,引入了电子,N掺杂石墨烯体系的费米能级移到导带区域,成为n型掺杂,体系表现出金属性,因此N掺杂提高了体系的导电性。Al-graphene体系的费米能级向下移到价带区域,Al掺杂石墨烯具有金属性。值得注意的是P掺杂会产生一个1.0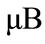 的磁矩,可以被用作具有半导体特性和磁性的特殊材料。
的磁矩,可以被用作具有半导体特性和磁性的特殊材料。
3.2. Si在替位掺杂石墨烯上的吸附
Si在N掺杂石墨烯上吸附结构如图3(a)所示,Si的稳定吸附位置为N-C键的中点。由于Si原子与N掺杂体系的相互作用,N原子向上凸起了0.324 Å,Si在N掺杂石墨烯上吸附的形成能为1.037 eV(表1),与空位和B掺杂石墨烯[15]相比,N掺杂对Si在石墨烯上吸附的影响很小。对于Al-graphene体系,Si的稳定吸附位置为Al的正上方(如图3(b)),由于Si原子与Al-graphene体系的相互作用,Al原子向下移动了1.198 Å,Si吸附Al-graphene体系的形成能为7.691 eV,相比其它体系形成能最大。而对于P-graphene体系,Si原子吸附在最近邻碳原子顶位(如图3(c)),Si吸附高度为2.644 Å,P向上凸起1.050 Å,其形成能为2.652 eV,对Si吸附的作用弱于Al-graphene体系而强于N-graphene体系。总之,石墨烯掺杂不同原子后,不同的体系对Si原子稳定吸附的位置不相同,而对于Si原子的吸附能力也有差别,其中Al、P掺杂增强了Si在石墨烯上的吸附,N掺杂对Si在石墨烯上吸附的影响很小。
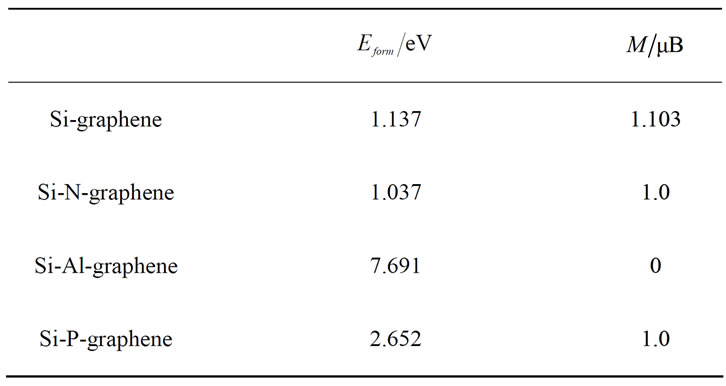
Table 1. Formation energy (Eform) and magnetic moment (M) of Si adsorbed on different grapheme systems
表1. Si在不同石墨烯体系上吸附的形成能和磁矩
 (a) (b) (c)
(a) (b) (c)
Figure 3. Top views and side views of Si adsorbed on N-doped (a), Al-doped (b), and P-doped (c) graphene. The small black ball represents the C atoms, the big black ball represents the Si atom, the big gray ball represents the Al atom, the dark gray ball represents the N atom and the small light gray ball represent P atom, respectively
图3. Si原子吸附在N掺杂(a)、Al掺杂(b)和P掺杂(c)石墨烯体系上的俯视与侧视结构图,其中黑色小球为C原子,黑色大球为Si原子,灰色大球为Al原子,深灰色与浅灰色小球分别为N和P原子

Figure 4. Density of states (DOS)for Si adsorbed on doped graphenesystems. (a) Si-graphene structure, (b) Si-N-graphene structure, (c) Si-Al-graphene structure, (d) Si-P-graphene structure. The solid curve shows the DOS of doped grapheme systems, and the vertical dotted line represents the Fermi level
图4. Si吸附在掺杂各体系上的DOS图:(a) Si-graphene结构体系、(b) Si-N-graphene结构体系、(c) Si-Al-graphene结构体系、(d) Si-P-graphene结构体系。图中实线表示掺杂体系DOS,虚线表示体系的费米能级
Si吸附在掺杂石墨烯体系DOS如图4,对于Si-graphene体系,Si吸附后改变了衬底的态密度分布,原来全空的导带变为部分被占据,费米能级附近明显出现了两个峰,体系表现出金属性,自旋性质发生改变,体系显示磁性,磁矩为1.103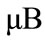 。对于N-graphene体系,Si吸附在N掺杂石墨烯体系后产生了自旋极化,自旋向上与自旋向下态密度不再对称,自旋性质发生了改变,体系显示磁性,磁矩为1.0
。对于N-graphene体系,Si吸附在N掺杂石墨烯体系后产生了自旋极化,自旋向上与自旋向下态密度不再对称,自旋性质发生了改变,体系显示磁性,磁矩为1.0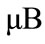 。这主要是由于Si的p轨道电子引起的。对于Al-graphene体系,Si吸附后,原来全空带的导带变为部分占据,自旋向上与自旋向下的态密度对称,因此体系不显磁性。对于P-graphene体系,Si吸附后,费米能级向下移动到价带区域,自旋向下与自旋向上的态密度不再对称,因而体系具有磁矩,磁矩大小为1.0
。这主要是由于Si的p轨道电子引起的。对于Al-graphene体系,Si吸附后,原来全空带的导带变为部分占据,自旋向上与自旋向下的态密度对称,因此体系不显磁性。对于P-graphene体系,Si吸附后,费米能级向下移动到价带区域,自旋向下与自旋向上的态密度不再对称,因而体系具有磁矩,磁矩大小为1.0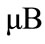 。
。
4. 结论
本文用第一原理计算方法研究了N、Al和P掺杂对Si在石墨烯上吸附的影响,结果表明:N原子与衬底C原子在同一平面内的N掺杂石墨烯为n型掺杂,N掺杂提高了体系的导电性;Al、P掺杂石墨烯引起石墨烯表面形成凸起,N、P原子化学掺杂石墨烯比Al掺杂石墨烯容易;Al掺杂石墨烯具有金属性,P掺杂会产生一个1 的磁矩;Si在N、Al、P掺杂石墨烯的稳定吸附位置分别为N-C键的中点、Al原子的正上方和P原子最近邻的碳原子顶位;其中Al、P掺杂增强了Si在石墨烯上的吸附,N掺杂对Si在石墨烯上吸附的影响很小;Si吸附N-graphene、P-graphene体系具有磁性,Si吸附Al-graphene体系磁矩为零,不显示磁性。
的磁矩;Si在N、Al、P掺杂石墨烯的稳定吸附位置分别为N-C键的中点、Al原子的正上方和P原子最近邻的碳原子顶位;其中Al、P掺杂增强了Si在石墨烯上的吸附,N掺杂对Si在石墨烯上吸附的影响很小;Si吸附N-graphene、P-graphene体系具有磁性,Si吸附Al-graphene体系磁矩为零,不显示磁性。
5. 致谢
本文感谢国家自然科学基金(11047014)以及北京计算科学研究中心的支持。感谢河南师范大学物理与信息工程学院与徐州师范大学理论物理研究所的老师和同学们对于撰写本论文所提供的帮助。
参考文献 (References)
[1] K. S. Novoselov, A. K. Geim, S. V. Mozorov, et al. Electric field effect in atomically thin carbon films. Science, 2004, 306(5996): 666-669.
[2] A. K. Geim, S. V. Mozorov. The rise of grapheme. Nature Materials, 2007, 6(3): 183-191.
[3] D. Garcia-Sanchez, A. M. van der Zande, A. San Paulo, et al. Imaging mechanical vibrations in suspended graphene sheets. Nano Letters, 2008, 8(5): 1399-1403.
[4] J. Moser, A. Verdaguer, D. Jiménez, et al. The environment of graphene probed by electrostatic force microscopy. Applied Physics Letters, 2008, 92(12): Article ID 123507.
[5] G. Giovannetti, P. A. Khomyakov, G. Brocks, et al. Doping graphene with metal contacts. Physical Review Letters, 2008, 101(2): Article ID 026803.
[6] K. T. Chan, J. B. Neaton and M. L. Cohen. First-principles study of metal adatom adsorption on Grapheme. Physical Review B, 2008, 77(23): Article ID 235430.
[7] H. Sevincli, M. Topsakal, E. Durgun, et al. Electronic and magnetic properties of 3d transition-metal atom adsorbed graphene and graphene nanoribbons. Physical Review B, 2008, 77(19): Article ID 195434.
[8] M. Wu, E.-Z. Liu and J. Z. Jiang. Magnetic behavior of graphene absorbed with N, O, and F atoms: A first-principles study. Applied Physics Letters, 2008, 93(8): Article ID 082504.
[9] Y. G. Zhou, X. T. Zu, F. Gao, et al. Adsorption-induced magnetic properties and metallic behavior of grapheme. Applied Physics Letters, 2009, 95(12): Article ID 123119.
[10] M. Endo, T. Hayashi, S.-H. Hong, et al. Scanning tunneling microscope study of boron-doped highly oriented pyrolytic graphite. Journal of Applied Physics, 2001, 90(11): 5670-5674.
[11] R. Czerw, M. Terrones, J.-C. Charlier, et al. Identification of electron donor states in N-doped carbon nanotubes. Nano Letters, 2001, 1(9): 457-460.
[12] P. Chen, X. Wu and J. Lin. High H2 uptake by alkali-doped carbon nanotubes under ambient pressure and moderate temperatures. Science, 1999, 285(5424): 91-93.
[13] V. Barone, J. E. Peralta, J. Uddin, et al. Screened exchange hybrid density-functional study of the work function of pristine and doped single-walled carbon nanotubes. The Journal of Chemical Physics, 2006, 124(2): Article ID 024709.
[14] S. Lattil, S. Roche, D. Mayyou, et al. Mesoscopic transport in chemically doped carbon nanotubes. Physical Review Letters, 2004, 92(25): Article ID 256805.
[15] 戴宪起, 李艳慧, 赵建华等. 空位和B掺杂对Si在石墨烯上吸附的影响[J]. 物理化学学报, 27(2): 369-373.
[16] G. Kresse, J. Hafner. Norm-conserving and ultrasoft pseudopotentials for first-row and transition elements. Journal of Physics: Condensed Matter, 1994, 6(40): 8245-8257.
[17] J. P. Perdew, K. Burke and M. Ernzerhof. Generalized gradient approximation made simple. Physics Review Letters, 1996, 77(18): 3865-3868.
[18] J. Y. Dai, J. M. Yuan and P. Giannozzi. Gas adsorption on graphene doped with B, N, Al, and S: A theoretical study. Applied Physics Letters, 2009, 95 (23): Article ID 232105.
[19] P. A. Denis. Band gap opening of monolayer and bilayer graphene doped with aluminium, silicon, phosphorus, and sulfur. Chemical Physics Letters, 2010, 492(4-6): 251-257.