Hans Journal of Computational Biology
Vol.4 No.01(2014), Article ID:13271,10 pages
DOI:10.12677/HJCB.2014.41001
Prediction of Four Kinds of Supersecondary Structures in Enzymes by Using Ensemble Classifier Based on SVM
College of Sciences, Inner Mongolia University of Technology, Huhhot
Email: *hxz@imut.edu.cn
Copyright © 2014 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received: Feb. 28th, 2014; revised: Mar. 7th, 2014; accepted: Mar. 11th, 2014
ABSTRACT
Enzymes are a kind of protein that has catalytic function. The study of supersecondary structures in enzymes plays an important role in the structure and function of enzymes. Based on enzyme sequence information, four kinds of supersecondary structures in enzymes were researched for the first time. Amino acids of sites and dipeptide components of sites were selected as parameters, for five selections of the best fixed-length pattern, the predictive results in 7-fold cross-validation were not ideal by using scoring function method; scores were selected as input parameters of support vector machine (SVM); the results were fused with weighted factors by using ensemble classifier; the better performance was obtained; the overall prediction accuracy was 72.64% and the Matthews correlation coefficient was above 0.57. Therefore, ensemble classifier based on SVM is an effective method to predict four kinds of supersecondary structures in enzymes.
Keywords:Enzyme; Supersecondary Structure; Scoring Function; Support Vector Machine; Ensemble Classifier

基于支持向量机的整体分类器算法
预测酶蛋白质中四类简单超二级结构
高苏娟,胡秀珍
内蒙古工业大学理学院,呼和浩特
Email: *hxz@imut.edu.cn
收稿日期:2014年2月28日;修回日期:2014年3月7日;录用日期:2014年3月11日

摘 要
酶是一种具有催化功能的蛋白质,研究酶蛋白质中的超二级结构对研究酶的结构及功能有重要作用。本文从酶蛋白质序列出发,首次对酶蛋白质中的四类简单超二级结构进行研究。以位点氨基酸及其紧邻关联为参数,选取五种序列片段截取方式,采用7-交叉检验,使用矩阵打分方法预测的结果不理想;将矩阵打分值作为特征参数输入支持向量机,并用整体分类器进行加权融合,得到了较好的预测结果,预测总精度达到72.64%,Matthew’s相关系数在0.57以上,因此,基于支持向量机的整体分类器方法是一种有效的预测酶蛋白质中超二级结构的方法。
关键词
酶蛋白质;超二级结构;矩阵打分;支持向量机;整体分类器

1. 引言
酶是活细胞内产生的具有高度专一性和催化效率的蛋白质,又称为生物催化剂,生命活动中引起新陈代谢的千千万万的化学变化几乎都是在酶的催化下进行的,酶与生命现象息息相关。因此,对于酶结构及功能的研究对生命科学的发展至关重要。近年来研究者们在酶蛋白质分子的功能研究上获得了较大的成果,比如关于酶与非酶[1] [2] 、酶的亚类分类[3] -[7] 方面的研究。但是对酶蛋白质结构的研究还相对较少,只有2011年Liu和Hu[8] 、2012年Long和Hu[9] 对酶蛋白质中的β发夹模体进行了识别。
酶作为一种具有催化功能的蛋白质,它具有一般蛋白质分子所有的一级结构和高级结构,蛋白质的超二级结构(supersecondary structure)是指两个或几个二级结构单元被连接多肽(loop)连接起来,进一步组成有特殊几何排列的局域空间结构,简称Motif[10] 。简单超二级结构分为β-loop-β、β-loop-α、α-loop-α和α-loop-β四类。由于超二级结构是α螺旋、β折叠简单排列形成的局域结构,有着比较强的序列信号,而且在三级结构中频繁出现,对蛋白质折叠及稳定性起重要作用,因此,学者们非常重视对超二级结构的研究,做了许多工作[11] -[18] 。酶蛋白质中的超二级结构除了具有一般蛋白质中超二级结构的特点,还有其自身特点,常常参与形成一些结合位点和活性位点,执行复杂的生物学功能。例如,丝裂原活化蛋白激酶(mitogen-activated protein kinases, MAPKs)是信号从细胞表面传导到细胞核内部的重要传递者,其中就包含一个β-loop-α结构,氨基端的β折叠和羧基端的α螺旋之间形成一个裂隙,为ATP结合位点[19] 。又如,SnRK3是植物特有的一类蛋白激酶,又被称为类钙调磷酸酶B亚基互作蛋白激酶(calcineurin B-like calcium sensor-interacting protein kinases, CIPK)。CIPK激酶在C端的酶结合区中含有一个抑制区域,与钙离子结合蛋白CBL(calcineurin B-like calcium sensor, CBL)结合来激活这些激酶。而CBL蛋白有包含4个α-loop-α结构的保守核心区域[20] ,每个α-loop-α结构的保守性与结合的激酶的差异有关。因此,酶蛋白质中的简单超二级结构对酶结构及功能研究有特殊意义。
对蛋白质中四类超二级结构的研究,2008年Hu和Li[16] 、2010年Zou[15] 等人取得了较好的预测结果。本文在前人研究各类蛋白质超二级结构的基础上,首次对酶蛋白质中的简单超二级结构进行研究,将2261个酶蛋白质的超二级结构,按照loop连接的二级结构类型,分为β-loop-β、β-loop-α、α-loop-α和α-loop-β四类。从超二级结构的一级序列出发,序列模式固定长度选取24个氨基酸残基,采用第6位点为loop的N端、第19位点为loop的C端、第10位点为loop的N端、第15位点为loop的C端和以loop序列为中心对齐五种片段截取方式,以位点氨基酸和位点氨基酸紧邻关联作为参数,分别采用矩阵打分算法和支持向量机方法的预测结果不理想,将支持向量机的预测结果通过整体分类器加权融合,进一步预测四类超二级结构,取得了较好的预测效果。
2. 材料和方法
2.1. 数据库的构建及统计分析
选取SCOP (http://scop.mrc-lmb.cam.ac.uk/scop/)数据库中ASTRAL 1.75版的序列相似性≤95%的16712个蛋白质,从中删除一些小蛋白质后,剩余14977个蛋白质。经过Blastcluster软件处理,得到序列相似性<25%的蛋白质有8704个,其中,序列片段长度大于100个氨基酸残基、分辨率<3.0 Å的蛋白质有4442个。再从这4442个蛋白质中按照酶的EC编号[21] 挑选出2261个酶蛋白质(其中包括氧化还原酶393个,转移酶637个,水解酶776个,裂解酶199个,异构酶112个、连接酶115个和同时属于两种以上的酶29个)。根据dssp数据库提供的二级结构,将H、G、I归为α螺旋,E、B归为β折叠,其余为loop。按照loop连接的二级结构类型,得到独立的超二级结构单元53367个,其中,β-loop-β(以下用“EE”表示)有14037个,β-loop-α(以下用“EH”表示)有13391个,α-loop-α(以下用“HH”表示 )有13539个,α-loop-β(以下用“HE”表示)有12400个。对四类超二级结构序列片段进行统计分析,我们发现,loop长度主要集中在2-12个氨基酸之间(见图1),包含四类超二级结构单元45506个,其中,EE有12956个,EH有10646个,HH有10682个,HE有11222个,分别占总数的92.3%、79.5%、90.5%、78.9%。loop长度在2~12个氨基酸之间的四类超二级结构中,序列片段长度主要分布在6-30个氨基酸之间(见图2),包含四类超二级结构41793个,具体有EE12847个、EH10090个、HH8103个、HE10753个,分别占其中的99.2%、94.8%、75.8%、95.8%。因此,我们以loop长度在2~12个氨基酸之间的序列片段长度在6-30个氨基酸之间的超二级结构为研究对象。
2.2. 计算方法
2.2.1. 四类超二级结构序列片段的截取
通过对四类超二级结构序列片段的统计分析,我们得到EE、EH、HH、HE的平均长度分别为15个氨基酸、19个氨基酸、24个氨基酸、19个氨基酸。而且,α螺旋的平均长度为9个氨基酸,loop的平均长度为4个氨基酸,β折叠的平均长度为5个氨基酸。因此,为了保证四类超二级结构的重要信息都不丢失,选取固定模式长度为24个氨基酸,保证loop两端连接的二级结构都能进入序列片段,同时,由于loop两端有较强的氨基酸保守性,比如,氨基酸G在loop两端出现较为频繁[10] ,所以,我们采用以第6位点作为loop的N端、以第19位点作为loop的C端、以第10位点作为loop的N端、以第15位点作为loop的C端和以loop序列为中心对齐五种片段截取方式,见图3。
2.2.2. 位点信息的统计分析及参数选取
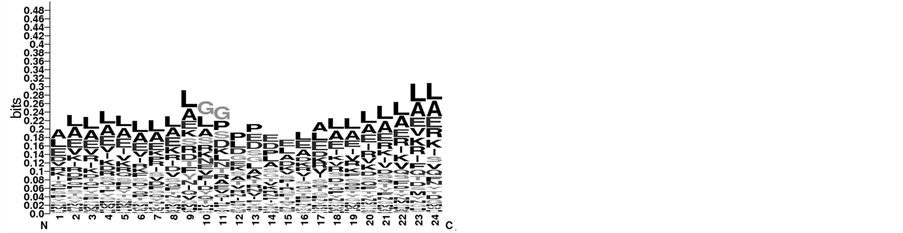
对2.2.1的5种序列片段截取方式,分别使用weblogo软件进行位点保守性统计分析,由于篇幅限制,这里选取部分为例说明(见图4)。以第10位点为loop的N端为例,(a) 图代表超二级结构EE,(b) 图代表超二级结构HH,比较(a)和(b),(a) 图中第10位点到第13位点最保守的氨基酸都是G,其中第10位点和第11位点氨基酸D的保守性次之,其它位点最保守的氨基酸多为V、L;而(b)图中第12、13位点最保守的氨基酸是P,第10位点和第11位点最保守的氨基酸虽然也是G,但是次之保守的氨基酸分别是L和P,其它位点最保守的氨基酸多为L、A。说明同一种片段截取方式,不同超二级结构的保守性不同。以超二级结构HE为例,(c) 图代表第15位点为loop的C端,(d) 图代表第6位点为loop的N端,
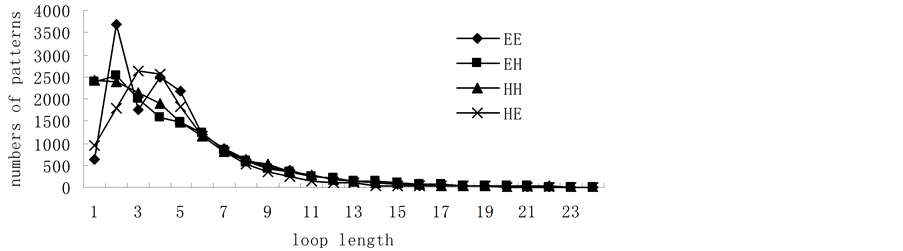
Figure 1. The distribution of sequence numbers with different loop length in the supersecondary structures
图1. 不同loop长度对应的四类超二级结构数目
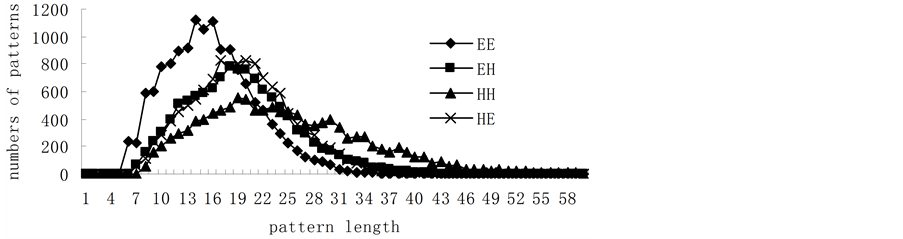
Figure 2. The distribution of pattern numbers with different pattern length
图2. 不同序列片段长度对应的四类超二级结构数目
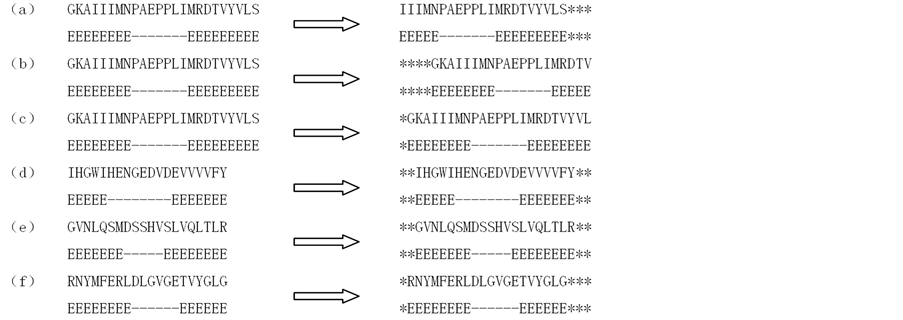
Figure 3. The diagram of the best patterns fixed-length: (a) beginning of loop locates the sixth position (b) end of loop locates the nineteenth position (c) beginning of loop locates the tenth position (d) end of loop locates the fifteenth position (e) loop sequence locates the center (the length of loop is an odd number) (f) loop sequence locates the center (the length of loop is a even)
图3. 最佳固定模式长度选取示意图:(a) 第6位点为loop N端;(b) 第19位点为loop C端;(c) 第10位点为loop N端;(d) 第15位点为loop C端;(e) 以loop序列为中心(loop长为奇数);(f) 以loop序列为中心(loop长为偶数)
Note: first row is amino acid sequences, second row is secondary structures corresponding sequences, “*”is a terminal residue
注:第一行表示氨基酸序列,第二行表示序列对应的二级结构,“*”表示一个空位
 (a)
(a)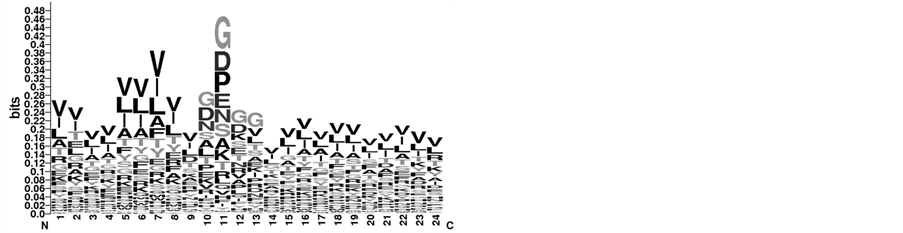 (b)
(b)
 (c)
(c) (d)
(d)
Figure 4. Sample of the position conservation: (a) beginning of loop locates the tenth position of EE (b) beginning of loop locates the tenth position of HH (c) end of loop locates the fifteenth position of HE (d) beginning of loop locates the sixth position of HE.
图4. 位点氨基酸的保守性举例:(a)以第10位点为loop的N端(EE) (b)以第10位点为loop的N端(HH) (c)以第15位点为loop的C端(HE)(d)以第6位点为loop的N端(HE)
Note: the overall height of the stack indicates the position conservation, while the height of symbols within the stack indicates the relative frequency of each amino acid at that position
比较(c)和(d),(c)图中第13、14位点最保守的氨基酸是G,第15位点最保守的氨基酸是P,其它位点最保守的氨基酸多为V、A、L;而(d)图中,第6、7、8位点最保守的氨基酸是G,其它位点最保守的氨基酸多为A、L、V,另外,第24位点的氨基酸保守性有明显特点,最保守的氨基酸是S、F、T、V。可见,不同的片段截取方式有着不同的位点氨基酸保守特性,所以5种截取方式的位点氨基酸保守信息可以作为预测参数。这里我们选取位点氨基酸(20种氨基酸加一个空格)和其紧邻关联为参数。
2.2.3. 矩阵打分算法(PCSF)
矩阵打分方法在转录因子结合位点预测方面取得较好结果[22] [23] 。本文以位点氨基酸及其紧邻关联的保守性作为参数,将酶蛋白质中的四类简单超二级结构用矩阵打分的方法分类。
矩阵相似性打分函数为: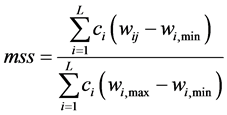
其中,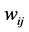 是位置权重矩阵的矩阵元,
是位置权重矩阵的矩阵元, ,
, 为选取的蛋白质超二级结构序列模式的片断长度,
为选取的蛋白质超二级结构序列模式的片断长度,
以氨基酸为参数的矩阵是21行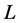 列,以氨基酸紧邻关联为参数的矩阵是441行
列,以氨基酸紧邻关联为参数的矩阵是441行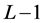 列。
列。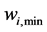 表示位置权重矩阵的第
表示位置权重矩阵的第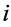 列上出现的矩阵元最小值,
列上出现的矩阵元最小值,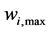 表示第
表示第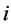 列上出现的矩阵元最大值。
列上出现的矩阵元最大值。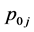 表示氨基酸
表示氨基酸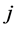 出现的背景概率。
出现的背景概率。
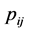 是位点位置概率,
是位点位置概率,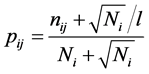 。以氨基酸为参数时,
。以氨基酸为参数时,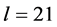 ,
,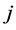 表示20种氨基酸和空位,
表示20种氨基酸和空位,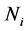
表示第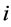 个位置上所有氨基酸出现的总数,
个位置上所有氨基酸出现的总数,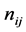 表示第
表示第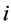 个位置上第
个位置上第 种氨基酸出现的频数;以氨基酸紧邻关联为参数时,
种氨基酸出现的频数;以氨基酸紧邻关联为参数时,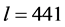 ,
, 表示第
表示第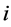 个位置上所有氨基酸紧邻关联出现的总数,
个位置上所有氨基酸紧邻关联出现的总数,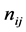 表示第
表示第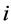 个位置上第
个位置上第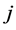 种氨基酸紧邻关联出现的频数。
种氨基酸紧邻关联出现的频数。
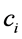 是位点保守性参量,反映被测序列在该位点与一致性序列在该位点的偏离性。
是位点保守性参量,反映被测序列在该位点与一致性序列在该位点的偏离性。
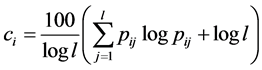
可以证明: ,如果等于1,则说明未知序列每一位点上的氨基酸刚好与位置权重矩阵位点上的最大值所对应的氨基酸一致。对含有24个氨基酸的固定序列模式片段,以位点氨基酸(位点氨基酸紧邻关联)作为参数,四类超二级结构可以构建4个21行24列(441行23列)的标准打分矩阵,任一条待测序列片段分别以4个标准位置打分矩阵为标准,利用打分函数,确定相似程度,得到4个打分值,哪一个分值最大,该序列就被预测为哪一类超二级结构。
,如果等于1,则说明未知序列每一位点上的氨基酸刚好与位置权重矩阵位点上的最大值所对应的氨基酸一致。对含有24个氨基酸的固定序列模式片段,以位点氨基酸(位点氨基酸紧邻关联)作为参数,四类超二级结构可以构建4个21行24列(441行23列)的标准打分矩阵,任一条待测序列片段分别以4个标准位置打分矩阵为标准,利用打分函数,确定相似程度,得到4个打分值,哪一个分值最大,该序列就被预测为哪一类超二级结构。
2.2.4. 支持向量机(SVM)方法
SVM是Vapnik[24] [25] 等人提出来的一类新型机器学习方法,目前已被成功地应用于蛋白质结构预测、蛋白质亚细胞定位及蛋白质折叠子的分类等多方面[26]
-[29] 。SVM的基本思想是基于统计学习理论,用核函数映射的方法把输入矢量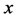 映射到一个高维特征空间,在高维特征空间构造出最优超平面,使得该超平面在保证分类精度的同时,能够使超平面两侧的空白区域最大化,从而达到最大的泛化能力,其由有限的训练集样本得到的小的误差能够保证对独立的检验集仍保持小的误差。常用的核函数有多项式核函数
映射到一个高维特征空间,在高维特征空间构造出最优超平面,使得该超平面在保证分类精度的同时,能够使超平面两侧的空白区域最大化,从而达到最大的泛化能力,其由有限的训练集样本得到的小的误差能够保证对独立的检验集仍保持小的误差。常用的核函数有多项式核函数 、径
、径
向基核函数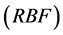 、
、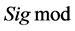 核函数
核函数 ,我们选择径向基核函数:
,我们选择径向基核函数: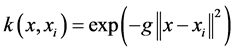 。另
。另
外,由于SVM算法是一个凸优化问题,局部最优解一定是全局最优解。因此SVM是一类很好的非线性模式识别分类器。SVM 算法已被很多学者编译成程序加以实现,常用的有libsvm、mysvm及svmlight等。这里我们使用的是libsvm-2.91程序包[30] ,其中c和gamma值为缺省。
根据2.2.3的矩阵打分算法,以位点氨基酸(位点氨基酸紧邻关联)为参数,对于训练集,四类超二级结构可以构建4个标准打分矩阵,用于检验集,可以得到4个打分值。将这些矩阵打分值作为特征参数输入支持向量机进行预测。
2.2.5. 整体分类器
整体分类器已用于预测27类蛋白质折叠子[31] ,取得了较好的预测效果。
定义整体分类器:
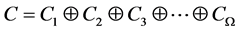 (1)
(1)
这里, 分别是基于支持向量机算法的
分别是基于支持向量机算法的 个单分类器,
个单分类器, 为分类器的融合。
为分类器的融合。
将 个单分类器
个单分类器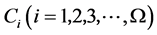 整合为整体分类器
整合为整体分类器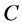 用于预测,可以由下式表示:
用于预测,可以由下式表示:
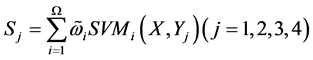 (2)
(2)
式中,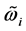 是单分类器
是单分类器 的权重系数,
的权重系数,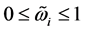 ,
,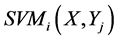 是任意一个待测序列
是任意一个待测序列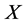 与单分类器
与单分类器 中第
中第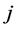 类标准源
类标准源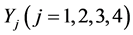 的支持向量机,
的支持向量机, 的最大值:
的最大值:
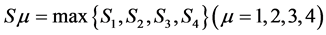 (3)
(3)
决定了待测序列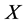 的预测类型。其中
的预测类型。其中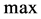 表示取最大值,
表示取最大值,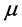 就是待测序列
就是待测序列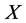 采用整体分类器预测所属的分类。
采用整体分类器预测所属的分类。
2.2.6. 系统检验
本文对分类结果的评价使用7交叉检验的方法,随机将数据库分为7个子集,依次取出1个子集作测试集,而其余6个子集作为训练集,此过程循环7次。
2.2.7. 精确度评价指标
定义第i类超二级结构(i = 1代表EE,i = 2代表EH,i = 3代表HH,i = 4代表HE)的敏感性指标Sni,特异性指标Spi,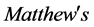 相关系数
相关系数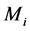 及预测精度S分别为:
及预测精度S分别为:
 (4)
(4)
 (5)
(5)
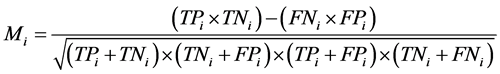 (6)
(6)
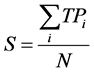 (7)
(7)
式中,TPi为该类中正确预测的样品数,FNi为该类中错误预测的样品数,TNi为其它类中正确预测的样品数,FPi为其他类被预测为此类的样品数,N表示四类序列总数。
3. 结果与讨论
3.1. 矩阵打分方法的计算结果与讨论
以位点氨基酸(21AA)和位点氨基酸紧邻关联(441JL)分别为参数,对5种序列片段截取方式:以第6位点为loop N端(N6);以第19位点为loop C端(C19);以第10位点为loop N端(N10);以第15位点为loop C端(C15);以loop序列为中心对齐(Center),用矩阵打分的方法预测酶蛋白质中四类超二级结构,7交叉检验的结果见表1。
从表中可以看出,21AA的个别结果较好,比如超二级结构EE中N6和C19的敏感性分别达到70.1%和77.2%,而超二级结构HH的计算结果很不理想,C19、N10、C15、Center的敏感性分别为17.7%、22.5%、25.1%、25.1%,Matthew’s相关系数0.1左右,超二级结构HE中N6的敏感性也仅为25%;441JL结果的HE中N6敏感性较好,达到79.2%,HH中C19的特异性、HE中N10的敏感性、EH中c15和center的敏感性都达到81.9%、81%、79.7%和80%,结果较好,HH的计算结果同样不理想,C19、N10、C15、Center的敏感性分别为25.5%、28.3%、24%、24.7%,Matthew’s相关系数0.27以上,C19的预测总精度为49.3%,结果较差。
3.2. SVM的计算结果与讨论
将矩阵打分值作为特征参数输入SVM,采用7-交叉检验,将位点氨基酸为参数的打分值输入SVM (21AA),每种片段截取方式的预测结果见表2。比较以位点氨基酸为参数的打分方法的预测结果(见表1中21AA),有些结果明显提高,比如,EE中N10、C15、Center截取方式的敏感性分别由打分方法的56%、56.9%、59.7%提高到现在的78.9%、76.4%、81.5%,HE中N6截取方式的敏感性由打分方法的25%提高到现在的67.4%,但是也有一些预测结果比打分方法的结果有所下降;将位点氨基酸紧邻关联为参数的打分值输入SVM (441JL),每种片段截取方式的预测结果(见表2)比较以同样参数的打分方法的预测结果(见表1中441JL)普遍有明显提高,比如,HH中N6、C19、N10、C15、Center截取方式的敏感性分别由打分方法的37.9%、25.5%、28.3%、24%、24.7%提高到现在的47.2%、50.7%、46.5%、51%、43.9%,

Table 1. The predictive results of PCSF algorithm using 7_Fold cross-validation
表1. 打分方法7交叉检验的预测结果
Table 2. The predicting results of SVM using 7_cross validation
表2. 支持向量机方法7交叉检验的预测结果
预测总精度分别由打分方法的54.3%、49.3%、55%、56.5%、58%提高到现在的61.8%、62.8%、62.2%、63.8%、61.9%。为了进一步提高预测精度,我们将SVM的计算结果输入整体分类器。
3.3. 整体分类器的计算结果与讨论
将位点氨基酸紧邻关联为参数的五种片段截取方式作为5个单分类器,采用整体分类器进行加权融合,进一步预测酶蛋白质中四类超二级结构。5个单分类器的支持向量机在不同权重系数下的预测结果见表3(篇幅所限,只列出代表性的结果,其它略去)。平均预测总精度最高达到72.64%,相关系数在0.57以上,相比表2中的预测结果,平均预测精度提高8.8个百分点以上,其它各项指标也显著提高。结果表明,整体分类器通过加权融合单分类器的计算结果,能有效的提取酶蛋白质中四类简单超二级结构中有益的预测信息,可以有效的提高预测精度。
目前,还没有预测酶蛋白质中四类简单超二级结构的相关文献,所以,我们只能参考前人对各类蛋白质中超二级结构的预测结果,见表4。2008年Hu和Li[16] 使用基于打分值和离散增量为组合向量的支持向量机识别算法对各类蛋白质中4类超二级结构进行分类,以氨基酸为参数,训练集的最高预测精度达到71.7%,与本文相比,除了EE的特异性,本文中所有指标的预测结果都优于Hu's。2010年Zou[15] 等人用SVM和IDQD的方法也对各类蛋白质中的四类简单超二级结构进行预测,其中IDQD方法的预测结果最好,以氨基酸组成、二肽组分和氨基酸组成分布共同为参数,训练集的最高预测精度达到77.7%,高于我们的预测总精度,但是,有些指标我们的算法高于Zou's,比如,我们的算法中,EH、HH、HE的特异性分别为78%、83%和74.3%,Matthew’s相关系数分别为0.63、0.65、0.64,而Zou’s算法中,EH、HH、HE的特异性分别为71%、73.3%和69.5%,Matthew’s相关系数分别为0.56、0.58、0.55。
4. 结论
酶蛋白质中的简单超二级结构对酶的生物学功能有重要影响,本文首次对酶蛋白质中四类简单超二级结构进行了理论预测。首先建立研究的数据集,我们的数据集包含四类超二级结构单元41793个,这个大数量的数据集帮助我们更加有效地预测酶蛋白质中超二级结构,是我们研究工作的有利基础。从酶
Table 3. The overall results for ensemble classifier
表3. 整体分类器的预测结果
Table 4. Comparison among different predictive results
表4. 不同方法预测结果的比较
序列出发,通过统计分析,选取最佳模式的长度为24个氨基酸,以位点氨基酸及其紧邻关联为参数,根据四类超二级结构的特点,采用五种片段截取方式,将计算出的矩阵打分值作为特征参数输入支持向量机,再通过整体分类器加权融合,得到较好的预测结果。
基于支持向量机算法的整体分类器能够有效的提高预测精度,首先,矩阵打分方法能降低参数的维数,而支持向量机算法能够融合有益的特征信息;其次,将各单分类器的计算结果进行加权融合可以提取更加有益的预测信息,从而提高预测的精度。本文只选择了位点氨基酸及其紧邻关联为参数,今后的工作中我们将考虑结合其它的有益参数,比如氨基酸的亲疏水性,柔性等等,也可以进一步调整分类器的个数和权重系数,进而获得更为理想的预测结果。
项目基金
国家自然科学基金资助项目(31260203, 30960090)。
参考文献 (References)
- [1] Cai, Y.D. and Chou, K.C. (2005) Using Functional Domain Composition To Predict Enzyme Family Classes. Journal of Proteome Research, 4, 109-111.
- [2] Cai, Y.D., Guo, P.Z. and Chou, K.C. (2005) Predicting Enzyme Family Classes by Hybridizing Gene Product Composition and Pseudo-Amino Acid Composition. Journal of Theoretical Biology, 234, 145-149.
- [3] Chou, K.C. and Cai, Y.D. (2004) Using GO-PseAA Predictor to Predict Enzyme Sub-Class. Biochemical and Biophysical Research Communications, 325, 506-507.
- [4] Shen, H.B. and Chou, K.C. (2007) EzyPred: A Top-Down Approach for Predicting Enzyme Functional Classes and Subclasses. Biochemical and Biophysical Research Communications, 364, 53-59.
- [5] Shi, R.J. and Hu, X.Z. (2010) Predicting Enzyme Subclasses by Using Support Vector Machine with Composite Vectors. Protein and Peptide Letters, 17, 599-604.
- [6] Hu, X.Z. and Ting, W. (2011) Prediction of Enzyme Subclass by Using Support Vector Machine Based on Improved Parameters. 2011 7th International Conference on Natural Computation, Shanghai, 26-28 July 2011, 593-598.
- [7] Wang, Y. and Hu, X.Z. (2011) Predicting of Oxidoreductase and Lyase Subclasses by Using Support Vector Machine. 2011 10th IEEE/ACIS International Conference on Computer and Information Science, Sanya, 16-18 May 2011, 27- 31.
- [8] Liu, X.X. and Hu, X.Z. (2011) Identifying the β-Hairpin Motifs in Enzymes by Using Support Vector Machine. 2011 10th IEEE/ACIS International Conference on Computer and Information Science, Sanya, 16-18 May 2011, 21-26.
- [9] Long, H.X. and Hu, X.Z. (2012) Prediction β-Hairpin Motifs in Enzyme Protein Using Three Methods. 2012 8th International Conference on Natural Computation (ICNC 2012), Chongqing, 29-31 May 2012, 570-574.
- [10] 阎隆飞, 孙之荣 (1999) 蛋白质分子结构.清华大学出版社, 北京, 43-56.
- [11] Kuhn, M., Meiler, J. and Baker, D. (2004) Strand-Loop-Strand Motifs: Prediction of Hairpin and Diverging Turns in Proteins. Protein, 5, 282-288.
- [12] Cruz, X., Hutchinson, E.G., Shepherd, A., et al. (2002) Predicting Protein Topology: An Approach to Identifying Bhairpins. Proceedings of the National Academy of Sciences, 99, ll157-1l162.
- [13] Kumar, M., Bhasin, M., Natt, N.K., et al. (2005) BhairPred: Prediction of β-Hairpins in a Protein from Multiple Alignment Information Using ANN and SVM Techniques. Nucleic Acids Research, 33, 154-159.
- [14] 胡秀珍, 李前忠 (2006) 用离散量的方法识别蛋白质的超二级结构. 生物物理学报, 6, 424-428.
- [15] Zou, D.S., He, Z.S., He, J.Y., et al. (2011) Supersecondary Structure Prediction Using Chou’s Pseudo Amino Acid Composition. Journal of Computational Chemistry, 32, 271-278.
- [16] Hu, X.Z. and Li, Q.Z. (2008) Prediction of the β-Hairpins in Proteins Using Support Vector Machine. The Protein Journal, 27, 115-122.
- [17] Hu, X.Z., Li, Q.Z. and Wang, C.L. (2010) Recognition of β-Hairpin Motifs in Proteins by Using the Composite Vector. Amino Acids, 38, 915-921.
- [18] Sun, L.X., Hu, X.Z. and Li, S.B. (2012) Predicting βαβ Motifs Based on SVM by Using the ID and MS Values. 2012 5th International Conference on BioMedical Engineering and Informatics (BMEI 2012), Chongqing, 16-18 October 2012, 910-914.
- [19] Wang, Z., Harkins, P.C., Ulevitch, R.J., Han, J.H., Cobb, M.H. and Goldsmith, E.J. (1997) The Structure of MitogenActivated Protein Kinase p38 at 2.1-Å Resolution. Proceedings of the National Academy of Sciences, 94, 2327-2332.
- [20] Batistic, O. and Kudla, J. (2004) Integration and Channeling of Calcium Signaling through the CBL Calcium Sensor/ CIPK Protein Kinase Network. Planta, 219, 915-924.
- [21] Webb, E.C. (1992) Enzyme Nomenclature. Academic Press, SanDiego.
- [22] Cartharius, K., Frech, K., Grote, K., et al. (2005) Mat Inspector and Beyond: Promoter Analysis Based on Transcription Factor Binding Sites. Bioinformatics, 21, 2933-2942.
- [23] Kel, A.E., GoBling, E., Reuter, I., et al. (2003) MATCHTM: A Tool for Searching Transcription Factor Binding Sites in DNA Sequences. Nucleic Acids Research, 31, 3576-3579.
- [24] Vapnik, V. (1995) The Nature of Statistical Learning Theory. Springer, New York.
- [25] Vapnik, V. (1998) Statistical Learning Theory. Wiley-Interscience, Hoboken
- [26] Hu, X.Z. and Li, Q.Z. (2008) Using Support Vector Machine to Predict β-Turns and γ-Turns in Proteins. Computational Chemistry, 29, 1867-1875.
- [27] Chou, K.C. and Cai, Y.D. (2002) Using Functional Domain Composition and Support Vector Machines for Prediction of Protein Subcellular Location. Journal of Biological Chemistry, 227, 45765-45769.
- [28] Ding, C.H.Q. and Dubchak, I. (2001) Multi-Class Protein Fold Recognition Using Support Vector Machines and Neural Networks. Bioinformatics, 17, 349-358.
- [29] Shi, J.Y., Pan, Z., Zhang, S.W. and Liang, Y. (2006) Protein Fold Recognition with Support Vector Machines Fusion Network. Progress in Biochemistry Biophysics, 3, 155-162.
- [30] Chang, C.C. and Lin, C.J. (2001) LIBSVM: A Library for Support
Vector Machines. Software.
http://www.Csie.ntu.edu.tw/cjlin/libsvm

NOTES
*通讯作者。