Pharmacy Information
Vol.05 No.02(2016), Article ID:17464,5
pages
10.12677/PI.2016.52002
Syntheses of Impurity A and C of Olmesartan Medoxomil
Weisi Li, Guoping Chen, Xin Fan, Jingyu Tang
Jiangsu Zhongbang Pharmaceutical Co., Ltd., Nanjing Jiangsu

Received: Mar. 30th, 2016; accepted: Apr. 26th, 2016; published: Apr. 29th, 2016
Copyright © 2016 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



ABSTRACT
Objective: A synthetic method towards impurity A and C of olmesartan medoxomil has been provided through optimizing literature reaction conditions. Methods: Impurity A was directly obtained by acidic-removal of Trt protecting group of starting material 1 with subsequent recrystallization from acetone, whereas impurity C was afforded through dehydration of tertiary alcohol group of starting material 2 followed by acidic-removal of Trt protecting group. Results: Through optimization of literature reaction condition, impurity A and C with chemical purity greater than 95% can be readily obtained. Conclusion: The current method towards impurity A and C is simple and practical with good reproductivity.
Keywords:Olmesartan Medoxomil, Impurity A and C, Syntheses

奥美沙坦酯杂质A和C的合成
李维思,陈国萍,范鑫,唐景玉
江苏中邦制药有限公司,江苏 南京

收稿日期:2016年3月30日;录用日期:2016年4月26日;发布日期:2016年4月29日

摘 要
目的:优化和改进奥美沙坦酯杂质A和C的合成反应条件,提供了一条简单实用的合成奥美沙坦酯杂质A和C杂质对照品的路线。方法:杂质A由原料1通过一步酸解脱除三苯甲基保护基,然后用丙酮重结晶得到,而杂质C则通过原料2经叔醇官能团脱水成双键和酸解脱除三苯甲基保护基两步反应,最后通过柱层析得到。结果:优化了反应条件,重现性好,纯度大于95%符合杂质对照品要求。结论:本方法反应方法操作简单实用,重现性好。
关键词 :奥美沙坦酯,杂质A和C,合成

1. 引言
奥美沙坦酯是一种新型的经FDA批准上市的血管紧张素II受体阻滞剂,主要用于治疗高血压,具有半衰期长、不良反应少、疗效显著等优点 [1] - [3] 。但是其在工艺生产和储存过程中,会产生一些杂质。杂质的存在直接或间接的影响着药品的纯度和质量,因此,为保证药品质量和患者用药安全,就需要对奥美沙坦酯的杂质进行定量和定性的研究 [4] - [8] 。奥美沙坦酯活性成分中可能存在的杂质A-F如图1所示。本文主要研究杂质A和C的合成路线,为该仿制药的药物质量研究提供符合纯度要求的杂质标准对照品。杂质A的合成由起始原料1经一步酸解脱除三苯甲基保护基得到,而杂质C则通过原料2经叔醇脱水和酸解去除保护基得到(具体路线见图2和图3)。
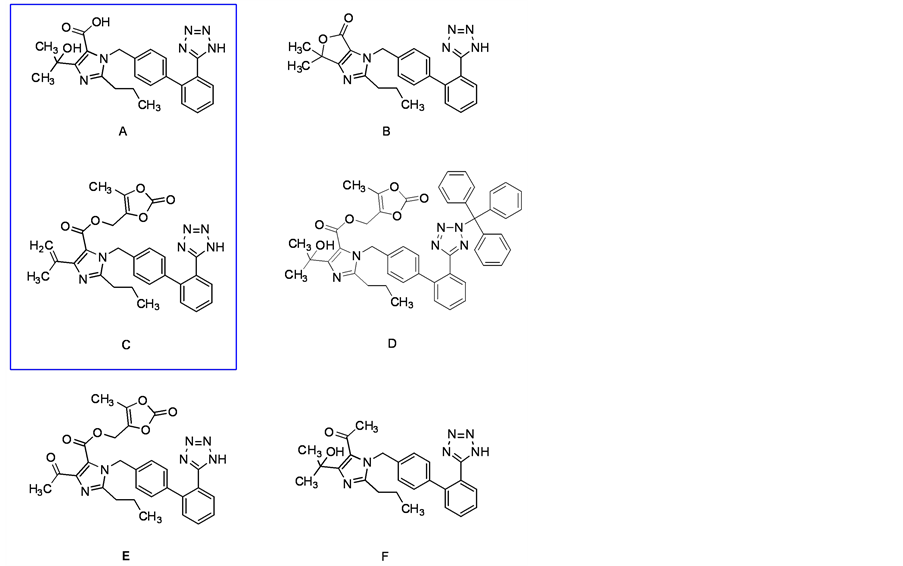
Figure 1. Impurity A-F of Olmesartan medoxomil
图1. 奥美沙坦酯杂质A-F
 原料1:4-(1-羟基-1-甲基乙基)-2-丙基-1-[[2'-(1-三苯甲基-1H-四唑-5-基)-联苯-4-基]甲基]-1H-咪唑-5-羧酸
原料1:4-(1-羟基-1-甲基乙基)-2-丙基-1-[[2'-(1-三苯甲基-1H-四唑-5-基)-联苯-4-基]甲基]-1H-咪唑-5-羧酸
Figure 2. Synthesis of impurity A
图2. 杂质A的合成

 原料2:4-(1-羟基-1-甲基乙基)-2-丙基-1-[[2'-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲]-1H-咪唑-5-羧酸(5-甲基-2-氧代-1,3-二氧杂环戊烯-4-基)甲酯
原料2:4-(1-羟基-1-甲基乙基)-2-丙基-1-[[2'-(1-三苯甲基-1H-四唑-5-基)联苯-4-基]甲]-1H-咪唑-5-羧酸(5-甲基-2-氧代-1,3-二氧杂环戊烯-4-基)甲酯
Figure 3. Synthesis of impurity C
图3. 杂质C的合成
2. 实验部分
2.1. 仪器和试剂
集热式恒温加热磁力搅拌器(南京文尔设备有限公司),磁力搅拌器(南京文尔仪器设备有限公司),旋转蒸发仪(上海申胜生物技术有限公司),电热恒温鼓风干燥箱(上海精宏实验设备有限公司),核磁共振仪 Bruker AV300 (内标TMS) (BRUKER公司),质谱 AGILENT 1200 LC-MSD (安捷伦公司),试剂为分析纯,使用前未进一步纯化。
2.2. 合成方法
2.2.1. 杂质A:4-(1-羟基-1-甲基乙基)-2-丙基-1-[[2'-(1H-四唑-5-基)-联苯-4-基]甲基]-1H-咪唑-5-羧酸 的合成(图2)
取20 mL 75%乙酸水溶液,加入原料(11.0 g, 1.45 mmol, Mw = 688.84),升温至50℃反应1小时。停止反应,将反应液冷却至室温,有白色晶体析出。抽滤,将滤液旋蒸得粘稠油状液体,加入20 mL水,用饱和碳酸氢钠调节pH值至6~7,抽滤得浅黄色固体,用大量二氯甲烷洗涤,最后用丙酮重结晶得产物0.43 g白色粉末,产率约66%。HPLC:96.8%。
红外:3420,2970,1636,1462~1572,1364~1431,1336,1051~1190。
氢谱:1H NMR (300 MHz, DMSO) δ:7.66 (m, 2H),7.57 (m, 2H),7.08 (m, 2H),6.97 (m, 2H),5.66 (s, 2H),2.58 (m, 2H),1.54 (m, 8H),0.85 (m, 3H)。
碳谱:13C NMR (300 MHz, DMSO) δ 160.9,155.0,152.7,150.5,141.0,138.1,136.8,130.9,130.5,129.0,127.7,125.9,123.4,118.1,70.6,47.0,29.6,27.9,20.4,13.5。
质谱:ESI/MS [M + 1]:447.2,ESI/MS [M + 1]:445.2。
2.2.2. 中间体1:4-(1-甲基乙烯基)-2-丙基-1-((2'-(1-三苯甲基-1H-四唑-5-基)-[1,1'-联苯]-4-基)甲基)-1H-咪唑-5-羧酸(5-甲基-2-氧代-1,3-二氧杂环戊烯-4-基)甲酯的合成(图3)
在100 mL的三颈圆底烧瓶中加入原料2 (2 g, 2.50 mmol),50 mL甲苯和0.1 g对甲苯磺酸,回流反应9小时。取出反应瓶,瓶内液体呈棕色,内壁有油状物质附着,以二氯甲烷溶解,然后将液体旋干,得棕色泡沫状固体。柱层析分离提纯,洗脱剂为石油醚:乙酸乙酯 = 5:2,将样品用少量二氯甲烷溶解,采取湿法上样。最终得浅黄色胶状物0.8 g,产率为40%。
2.2.3. 杂质C:4-(1-甲基乙烯基)-2-丙基-1-[[2'-(1H-四唑-5-基)联苯-4-基]甲基]-1H-咪唑-5-羧酸(5-甲基-2-氧代-1,3-二氧杂环戊烯-4-基)甲酯的合成(图3)
在100 mL三颈圆底烧瓶中加入0.8 g中间体1和40 mL乙腈,将反应体系放在冰盐浴中冷却到−10℃,然后缓慢加入1.5 mL浓盐酸,加完后继续反应6小时。加入200 mL水,抽滤除去析出的白色粉末状固体,滤液加入220 mL乙酸乙酯,并用饱和碳酸氢钠调节pH值至6左右。分出有机相用饱和盐水洗,再用无水硫酸钠干燥,抽滤,旋干,得0.3 g白色固体粉末,产率70%。HPLC:97.7%。
红外:3417,2894~2966,1822,1709,1640,1466~1640,1372~1393,1227,1007~1134。
氢谱:1H NMR (300 MHz, DMSO) δ:7.59 (m, 4H),7.05 (m, 4H),5.46 (m, 2H),5.25 (m, 2H),5.04 (s, 2H),2.58 (m, 2H),2.02 (m, 6H),1.60 (m, 2H),0.89 (m, 3H)。
碳谱:13C NMR (300 MHz, DMSO) δ 159.7,155.0,152.0,151.7,148.7,141.0,140.2,138.2,137.7,136.4,133.1,130.9,130.5,129.0,127.7,125.8,123.4,117.0,53.6,47.3,28.1,22.1,20.3,13.5,8.7。
质谱:ESI/MS [M + 1]:541.3,ESI/MS [M + 1]:539.2。
3. 结果与讨论
参照奥美沙坦酯的合成 [1] ,杂质A中的三苯甲基保护基(Trt)选择乙酸水溶液中进行酸解脱除,是因为原料1和杂质A能溶于其中,而生成副产物乙酸三苯甲基酯不溶,通过简单抽滤即可分离除去。另外,Trt对乙酸比较敏感,在65℃~75℃下加热1小时就可以脱除干净,碱中和处理后从滤液中过滤得到的粗品外观带有浅黄色,HPLC测试显示纯度只有大约85%左右。但是我们发现用二氯甲烷进行打浆处理可以除去表面的颜色得到白色固体,然后经过2次丙酮重结晶后HPLC纯度可以提高到95%以上。
参照文献方法 [9] [10] ,杂质C的合成经由两步反应完成,第一步原料2以对苯甲磺酸为催化剂,通过甲苯共沸除水的方法来实现叔醇脱水到烯烃双键的转变,但实际实验结果显示反应很难进行完全,而且我们发现随着反应的进行,产生的中间体1将包裹原料2以及副反应产物以油状物的形式游离于反应溶液,并附着在瓶壁。因此,为了简化处理,反应结束后我们直接通过倾析的方式将反应液和油状物分离,然后用二氯溶解油状物进行柱层析分离得到中间体1。接着,杂质C的前体即中间体1是在−10℃温度下在浓盐酸/乙腈体系中脱去氨基上的Trt保护基,选择低温可以抑制中间体1上的酯基发生酸性水解的副反应。在该反应条件下,Trt保护基脱去后形成的副产物三苯甲基氯易溶于乙腈而微溶于水,而杂质C以盐酸盐的形式溶于水中,因此利用二者溶解度的差异可以将副产物三苯甲基氯与目标物杂质C进行有效分离。最后对反应液碱化中和,有机溶剂萃取可以直接得到符合杂质对照品药品的杂质C纯品。
4. 结论
本文通过优化和改进文献反应条件,提供了一种简单实用的奥美沙坦酯杂质A和C的合成方法。杂质A和C分别经一步和二步反应就可以获得,纯度大于95%,符合杂质对照品的技术要求。本文提供的方法后处理操作简单实用,重现性好,适合快速获取用于奥美沙坦酯质量研究的杂质A和C的对照品。
文章引用
李维思,陈国萍,范 鑫,唐景玉. 奥美沙坦酯杂质A和C的合成
Syntheses of Impurity A and C of Olmesartan Medoxomil[J]. 药物资讯, 2016, 05(02): 9-13. http://dx.doi.org/10.12677/PI.2016.52002
参考文献 (References)
- 1. Tao, X. and Liu, G.L. (2003) Olmesartan: A Novel Angiotensin II Receptor Antagonist. Chinese Journal of New Drugs and Clinical Remedies, 22, 368-370.
- 2. 张国天, 陈永生, 梁海军. 奥美沙坦治疗原发性高血压临床疗效分析[J]. 中国医院药学杂志, 2013, 33(11): 881-884.
- 3. 徐有恒, 林丽薇, 彭锋. 抗高血压新药—奥美沙坦酯[J]. 广东药学院学报, 2004, 20(3): 289-292.
- 4. 国家食品药品监督管理总局药品审评中心. 化学药物杂质研究技术指导原则[EB/OL]. http://www.cde.org.cn/zdyz.do?method=largePage&id=2060, 2007-08-23.
- 5. ICH (2006) Impurities in New Drug Substance Q3A(R2). http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q3A_R2/Step4/Q3A_R2__Guideline.pdf
- 6. ICH (2006) Impurities in New Drug Products Q3B(R2). http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q3B_R2/Step4/Q3B_R2__Guideline.pdf
- 7. EMEA (2012) Guideline on Setting Specifications for Related Impurities in Antibiotics. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129997.pdf
- 8. TGA (2013) Impurities in Drug Substances and Drug Products. https://www.tga.gov.au/guidance-18-impurities-drug-substances-and-drug-products
- 9. Fan, S.S., Zhao, L. and Zhang, Y. (2014) Synthesis of Related Substances of Olmesartan Medoxomil. Chinese Journal of Pharmaceuticals, 45, 1016-1018.
- 10. Karrothu, S.B., Amirisetty, R.T., Gade, S.R., et al. (2010) Synthesis of Related Substances of Olmesartan Medoxomil, Anti-Hypertensive Drug. Arkivoc, 2010, 292-302. http://dx.doi.org/10.3998/ark.5550190.0011.224