Hans Journal of Ophthalmology
Vol.2 No.2(2013), Article ID:11975,2 pages DOI:10.12677/HJO.2013.22002
A Case of Joubert Syndrome with Retinal Dysplasia
1Guangdong Medical College, Zhanjiang
2Department of Pediatrics, Affliated Hospital of Guangdong Medical College, Zhanjiang
Email: 847118482@qq.com, kenbobozj@yahoo.com.cn
Received: Mar. 5th, 2013; revised: Mar. 29th, 2013; accepted: Apr. 9th, 2013
Copyright © 2013 Linlin Wang, Yuge Huang. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT:
Joubert syndrome (JS) is a rare autosomal recessive congenital malformation of the brainstem and cerebellar vermis,which may be associated with other organ dysfunction (Joubert Syndrome and Related Disorders, JSRD), mainly as retinal dystrophy, nephronophthisis, hepatic fibrosis and polydactyly. It had been reported in the previous literature in our country, however, most do not mention other organ dysfunction. In the following case complicated with retinal dysplasia was discussed with relate literature.
Keywords: Joubert Syndrome; Retinal Hypoplasia; Cerebral Disease; Ophthalmology
Joubert综合征合并视网膜发育不良1例
王淋淋1,黄宇戈2
1广东医学院,湛江
2广东医学院附属医院儿科,湛江
Email: 847118482@qq.com, kenbobozj@yahoo.com.cn
摘 要:
Joubert综合征(Joubert syndrome, JS)是一种较为罕见的脑干及小脑蚓部先天性发育畸形,属于常染色体隐性遗传性疾病,该病可合并其它器官功能异常,即JS及相关畸形(Joubert Syndrome and Related Disorders, JSRD),如视网膜营养不良、肾囊肿、肝纤维化、多指畸形等[1]。在我国已有几例个案报道,但多未见合并其它器官异常,本例Joubert综合征有合并视网膜发育不良,本文结合相关文献进行讨论。
收稿日期:2013年3月5日;修回日期:2013年3月29日;录用日期:2013年4月9日
关键词:Joubert综合征;视网膜发育不良;脑部疾病;眼科学
1. 病例资料
患儿,男,4天,因“反应差、眼球震颤4天”入院。患儿为G6P4,胎龄38 + 2周,因其母为“高龄产妇,妊娠期糖尿病”于当地医院剖宫产出生,羊水清,脐带绕颈1周,无头颅血肿,无窒息抢救史,出生时Apgar评分1分钟为8分,予刺激足底、吸痰、吸氧等对症治疗,5分钟9分,10分钟不详,出生体重2.8 kg。生后反应差,呼吸促,故转入该院的新生儿科考虑为“新生儿肺炎,新生儿缺氧缺血性脑病”予吸氧、抗感染、营养脑神经等对症治疗4天后,患儿症状未见改善,病程中发现眼球水平震颤,不能凝视某物,多处于左斜视状态,故转入我院作进一步治疗。病程中患儿偶有四肢抖动,易惊跳,无发热、抽搐,奶粉喂养,奶量10 ml/次,有解胎便及小便,喜睡。其有一姐姐,为第3胎,现5岁,出生无窒息抢救史,生长发育迟缓,能走能跑,但不稳,语言能力仍欠佳,未发现有视力问题,未前往医院就诊过。母亲37岁,产6月发现脐带绕颈1周,孕35周因腹痛有保胎5天,未见胎心、胎动异常。
入院查体:精神反应稍差,营养中等,全身皮肤轻度黄染,前囟平软,颈无抵抗,无角弓反张,双眼不能盯某物,易震颤,多处于左斜视状态,双侧瞳孔等大等圆,直径约3 mm,对光反射灵敏,双肺呼吸音对症,双肺未闻及明显干湿罗音,心音有力,未闻及病理性杂音,腹部平软,未触及包块,脊柱无畸形,四肢活动自如,关节无异常,四肢肌张力阵发性增高,拥抱反射、吸吮反射、觅食反射、握持反射引出欠佳。
辅助检查:头颅MRI示:小脑体积缩小,小脑蚓部不全,双侧小脑半球间可见一“中线裂”,枕大池明显增宽并与增宽的第四脑室相互沟通,第四脑室上部扩大,呈“蝙蝠状”改变。DWI信号未见异常,小脑脚细小,呈“磨牙”状或齿征改变,双侧大脑半球灰白质及双基底节区信号正常,中线无移位,静脉窦留空效应正常。矢状位像见小脑与脑干近于垂直,考虑为Joubert综合征。泌尿系统及心脏彩超未见异常。听力筛查:右耳未通过。请眼科会诊行眼底检查提示:左眼视盘下有一类圆形视网膜病灶,考虑为视网膜发育不良。(如图1所示)。
根据病史、体征及检查,确诊为:1) Joubert综合征,2) 视网膜发育不良,于我科新生儿予脑营养治疗一疗程后,患儿气促、眼部震颤有改善,因本院眼科条件有限,转入上级医院作进一步诊疗。
2. 讨论
Joubert综合征是一种比较罕见的疾病,多表现为共济失调、呼吸急促、呼吸暂停、眼舌运动异常、肌张力减低等,主要由小脑蚓部发育不良或缺失及脑干缺陷造成,包括齿状核的变形、下橄榄核和旁橄榄核的发育不良、背侧柱核的异常和锥体交叉的几乎完全缺失[2]。于1969年由Joubert等首次报道5例小脑蚓部发育不全伴发作性呼吸过度、眼球活动异常、共济失调和智力低下的患儿[3]。Joubert综合征属于常染色体隐形遗传疾病,近年研究逐步发现与此病相关的基因突变位点,如C5ORF42[4]、INPP5E[5]、TMEM231[6]、C5ORF42[7]等,不同的人种及地域检测出不同的基因突变位点,为临床诊疗提供了充分的理论基础[8]。其
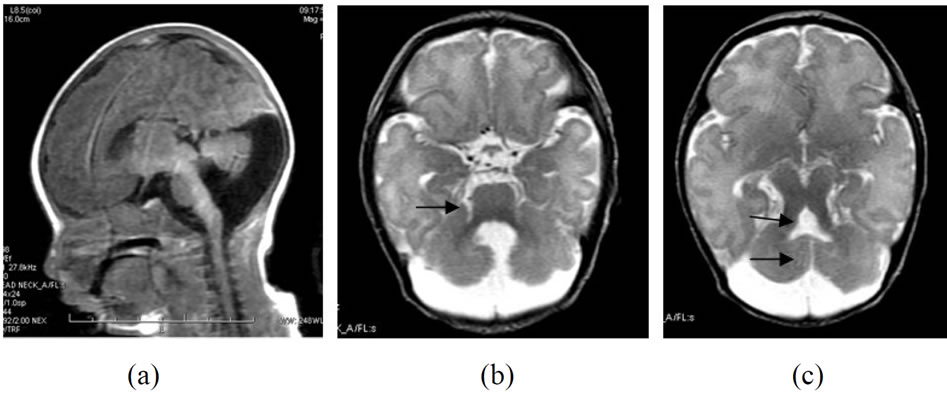
Figure 1. (a) Sagittal TIW image shows the cerebellum is perpendicular to the brain stem; (b) Axial T2 W image shows typical molar tooth appearance is seen with prominent superior cerebellar peduncles(black arrow); (c) Median clef is seen separating the cerebellar hemispheres and communicating with 4th ventricle,producing the typical bat wing (open umbrella) appearance
图1. (a) 矢状位像见小脑与脑干近于垂直;(b) 小脑脚细小,呈“磨牙”状或齿征改变;(c) 双侧小脑半球间可见一“中线裂”, 使枕大池与第四脑室相互沟通,第四脑室扩大,呈“蝙蝠状”或“雨伞状”改变
中合并视网膜发育不良的患儿,其染色体基因突变位点多为CEP290、AHI1[9,10],此例家属拒绝做染色体基因检测。
本病男性多见,Joubert综合征临床特征主要包括发作性呼吸过度和呼吸暂停、眼异常运动和儿童期发育迟缓,临床诊断通常在新生儿期。发作性呼吸过度和呼吸暂停,眼异常运动在新生儿期较明显,而发育迟缓在儿童期较明显,呼吸失调和眼异常运动在儿童期减少。眼部多表现为眼球水平震颤,可有视网膜发育不良或缺失,或脉络膜缺失,先天性失明,斜视,弱视等[11]。本例患儿为新生儿期,为男性,有呼吸急促,眼水平颤动等符合JS的临床表现。JS根据有无视网膜病变分为两种类型,一种是伴有视网膜发育不良,家族中有类似患者,另一种没有视网膜发育不良,家族中未发现类似患者,有文献报道该病近50%伴视网膜缺损和视网膜营养不良,而且视网膜发育不良的患者中30%有多囊肾病;该病15%以上患者有多指(趾)畸形。早期视网膜发育不良较难诊断,应定期进行眼的筛选检查才能发现。同样应定期肾功能和肾超声检查以发现囊性肾病,本例患儿有合并视网膜发育不良,且家族中有类似患者,但未发现合并肾脏畸形。
Joubert综合征影像学具有特征性,主要为小脑蚓部部分或完全缺乏、小脑上脚的增宽、第4脑室变形,这些表现几乎在所有的病例中均有存在。国外文献报道Joubert综合征影像学表现有“磨牙征”、“中线裂”、“蝙蝠翼状”和“雨伞状”的第4脑室[12]。“中线裂”是因蚓部的缺如,使正常的两小脑间缺乏正常蚓部,两侧小脑半球不连接,其间为细线状脑脊液相隔形成。小脑蚓部的完全缺如和几乎完全缺如,使颅侧和尾侧的第4脑室形成“蝙蝠翼”状和“雨伞状”。脚纤维束缺乏正常交叉,导致小脑上脚垂直地行走于中脑和小脑间的脑干中,致小脑上脚增宽,而且显得更为平行;小脑上脚的纤维缺乏交叉使中脑前后径缩短,尤其是中线区域,导致脚间池较正常更深。在横断面上增宽的中脑、凹陷加深的脚间池和平行状走行的小脑上脚在其周围脑脊液的衬托下,中脑和小脑上脚形态犹如磨牙的侧面观,被称为“磨牙征”。判断有无视网膜发育不良可通过眼底镜检查可直接观察到病变部位,视网膜电流图可见熄灭波形,还可通过眼电生理检查、眼底荧光血管造影等协助诊断,较大儿童还可进行视野检查,有些新生儿早期观察不到视网膜病变,可定期动态观察[13]。
Joubert综合征应与Dandy-Walker综合征、菱脑联合、Down综合征、橄榄球小脑萎缩等相鉴别。Dandy-Walker畸形也可伴小脑蚓部缺失,但其典型的表现是第四脑室向后上方扩张及颅后窝扩大,不仅可见小脑蚓部缺失,同样可见第4脑室从缺失的蚓部向后上方扩张,后颅凹异常大的囊性病变,小脑半球向前外方分离退缩,并通常伴发后颅凹的扩大,临床亦伴有发作性呼吸过度、动眼异常,但无“磨牙征”等MRI表现。菱脑联合的特点是两侧小脑半球之间无“中线裂”存在,小脑蚓部缺失、且两侧小脑半球呈融合改变,且无Joubert综合征特征性的临床表现可帮助鉴别。Down综合征仅凭影像学鉴别有困难,根据临床表现或染色体组型可明确诊断。橄榄球小脑萎缩(OPCA)、多系统萎缩–小脑共济失调型(MSA-C)、遗传性脊髓小脑共济失调(SCA),从临床上难以鉴别,可通过MRI特征性表现鉴别。该病合并的眼部病变应与斜视麻痹症(HGPPS)、牵牛花综合征、先天性脉络膜缺损、视乳头缺损和凹陷等疾病鉴别,此类病无MRI特征性表现,亦无肌张力减低、共济失调等临床表现,故容易相互鉴别。
参考文献 (References)
[1] V. Sturm, H. Leiba, M. N. Menke, et al. Ophthalmological findings in Joubert syndrome. Eye (Lond), 2010, 24(2): 222-225.
[2] M. A. Parisi. Clinical and molecular features of Joubert syndrome and related disorders. American Journal of Medical Genetics. Part C: Seminars in Medical Genetics, 2009, 151C(4): 326-340.
[3] M. Joubert, J. J. Eisenring, J. P. Robb, et al. Familial agenesis of the cerebellar vermis: A syndrome of episodic hyperpnea, abnormal eye movements, ataxia and retardation. Neurology, 1969, 19(9): 813-825.
[4] M. Srour, J. Schwartzentruber, F. F. Hamdan, et al. Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. American Journal of Human Genetics, 2012, 90(4): 693-700.
[5] L. Travaglini, F. Brancati, J. Silhavy, et al. Phenotypic spectrum and prevalence of INPP5E mutations in Joubert syndrome and related disorders. European Journal of Human Genetics, 2013.
[6] M. Srour, F. F Hamdan, J. A. Schwartzentruber, L. Patry, et al. Mutations in TMEM231 cause Joubert syndrome in French Canadians. Journal of Medical Genetics, 2012, 49(10): 636-641.
[7] M. Srour, J. Schwartzentruber, F. F. Hamdan, et al. Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. American Journal of Human Genetics, 2012, 90(4): 693-700.
[8] E. M. Valente, F. Brancati, E. Boltshauser, et al. Clinical utility gene card for: Joubert syndrome-update 2013. European Journal of Human Genetics, 2013.
[9] F. Brancati, G. Barrano, J. L. Silhavy, et al. CEP290 mutations are frequently identified in the oculo-renal form of Joubert’s syndrome-related disorders. American Journal of Human Genetics, 2007, 81(1): 104-113.
[10] M. A. Parisi, D. Doherty, M. L. Eckert, et al. AHI1 mutations cause both retinal dystrophy and renal cystic disease in Joubert’s syndrome. Journal of Medical Genetics, 2006, 43(4): 334-339.
[11] A. O. Khan, D. T. Oystreck, M. Z. Seidahmed, et al. Ophthalmic features of Joubert syndrome. Ophthalmology, 2008, 115(12): 2286-2289.
[12] P. Hurtado, H. Pachajoa. Molar tooth sign: A characteristic image in Joubert syndrome. Neurologia, 2010, 25(2): 140-141.