Journal of Advances in Physical Chemistry
Vol.2 No.4(2013), Article ID:12580,6 pages DOI:10.12677/JAPC.2013.24008
Quantum Chemical Study of the Role of the Van Der Waals Interaction (3)
China Zhongshan Yajule Saina Lake, Zhongshan
Email: 13923756124@163.com
Received: Aug. 3rd, 2013; revised: Sep. 18th, 2013; accepted: Sep. 25th, 2013
Copyright © 2013 Guangyao Zhou. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT:
In this article, the coupled-cluster method CCSD/aug-cc-pVTZ is employed to calculate a few van der Waals (vdW) systems, including dimer of hydrogen molecule, hydrogen-helium and hydrogen-neon complexes. The corresponding electronic density difference and graphics are also plotted. Based on the analysis of the graphs, it is suggested that the essence of intermolecular vdW interaction is the electron sharing between the interacting molecules under the repulsion and restriction of paired electrons, exhibiting the equilibrium between the repulsion and attraction of paired electrons, and showing directionality and saturation. vdW interaction is a kind of chemical effect accompanied by electron transfer, which satisfies equalization principle of electronegativity.
Keywords: Quantum Chemistry; Van Der Waals Interaction; Electron Density Difference; Hydrogen Molecules
范德华作用的量子化学研究(三)
周光耀
中国中山市雅居乐塞纳湖,中山
Email: 13923756124@163.com
摘 要:
本文用量子化学耦合簇CCSD/aug-cc-pVTz方法,计算了一些分子间范德华作用体系:氢分子二聚体,氢分子与氦、氖。并作了相应的电子密度差及图形。通过对图形分析,认为分子间范德华作用是在分子双方配对电子互斥、制约之下形成的电子共享,是配对电子互斥与电子共享吸引所形成的平衡,是有方向性和饱和性的,是一种化学作用。其中伴随着的电子转移,符合电负性均衡原理。
收稿日期:2013年8月3日;修回日期:2013年9月18日;录用日期:2013年9月25日
关键词:量子化学;范德华作用;电子密度差;氢分子
1. 引言
前文[1]说:量子化学计算,是在对于一个给定的原子核空间排布,如何安排电子使体系能量最低。而且通过化学体系的电子密度可以计算该体系的一切性质。反过来说该体系的一切性质变化必然通过电子密度变化反映出来。量子化学就是对大千世界化学物质的电子在核外运动、分布的一种数学模拟。
为了研究范德华作用,前文选择了稀有气体原子(Rg)来作为例子,因为这里形成分子时没有通常意义上的价键,只有范德华作用,可以从形成分子前后的电子密度变化,追寻范德华作用的足迹,揭示范德华作用的机制。
本文,则考查分子之间的范德华作用,选择氢分子作为例子,由于范德华力,氢分子能够形成聚合体,凝结成液体、成为结晶。
因为范德华作用如此之弱,它对氢分子的共价键影响微不足道,它对氢分子在聚合体中电子密度变化影响也微不足道,用氢分子形成的聚合体与各个自由氢分子作电子密度差,氢分子的电子密度就抵消了,只看到范德华力所形成聚合体前后电子密度的变化,从而继续追寻范德华作用的足迹,揭示范德华作用的机制。
本文用GAMESS-US程序[2],全部计算都用CCSD/aug-cc-pVTz进行,实践表明,该计算精确地计算了电子相关,能够正确地计算通常的成键和范德华作用。此外的计算方法则在文中说明。进一步用Multiwfn 3.1[3]进行数据处理。作图则用VMD、Sigmaplot等。
当距离数值不标出单位时是Å,电荷和能量数值单位是a.u.。
2. 量子化学计算和作图
2.1. 从氢分子说起
氢分子是单纯、典型的共价键,而且没有电荷在原子间转移,是本文讨论氢分子之间范德华作用的基础。
量子化学计算,就是从氢原子、氢分子起步逐渐发展起来的,新的量子化学计算方法,首先都要过算准氢原子、氢分子这道关。
计算的主要结果如下(括号内是实验值):
氢原子总能量:−0.4998212 (−0.4997278)。
氢分子键长:0.7426 (0.7416)。
氢分子总能量:−1.1726355 (−1.174447)。
氢分子结合能:−0.1729931 (−0.174)。
氢分子震动频率cm−1:4406.4 (4401.2)。
对于氢分子,无论从波函数图或电子云图看,它只是一个椭球。作它的电子密度差Δρ,才可以看出从自由氢原子形成氢分子前后电子变化的形象。
计算的各波函数文件wfn,在Multiwfn得到氢分子Δρ的cub文件。据此作图时,不能选择做等值面图,因为这样看见的只是一个负值的外壳,内部变化看不见。而要选择做截面等值线。见图1。这种图已是人所共知的了。
图中等值线的起始值取±0.0004。紫色为正,是电

Figure 1. H2 electron density differences
图1. 氢分子的电子密度差
子密度增加区域,增加电荷量记为+0.25038;青兰色为负,是电子密度减少区域,减少电荷量记为−0.25027。
从Δρ总体来看,电荷由四周向成键原子之间收缩、转移了。
由于二氢中间部位的电子受两个原子核共同吸引,这里的电子密度显著增加了,而周边密度减少了,特别是键轴方向的两边更加显著地减少了,电荷往中间移动了。形成了共享电子(图中显示的只是净增加的部分),这是共价键、有共价成分键的共同特征,在分子中具有普遍意义。
当然这种键轴上的密度增加,也是两个H的球形的电子云融合成一个椭圆球,需要填平二球之间的电子云不吻合、比较稀薄的部分,成为统一的椭圆球整体而形成的。
电子往键中间浓集引起H处电子减少,使得H处剩余电子因电子减少而受到核的吸引力有所增强,H处剩余电子会收缩,此处电子密度增加,所以在图1中,H处于电子浓集增加区,与键中间的电子增加融为一体了。
在氢分子中,虽然二H之间没有电荷转移,但电子却向键中间发生了聚集,二H处电荷总共减少了0.250,每个H减少的电荷为0.125。H原子仍然显著地失去了部分电子,电子对核屏蔽减弱,使得分子中的H电负性升高,能级降低。
我们通常一谈到原子、分子片之间成键,自然而然地就将新成键的能级,往下画一个比成键双方能级都低得多的能级,而不是二者的平均。这就是电子向中间移动,导致双方失去部分电子,能级降低的结果。
现在,我们不仅从计算得到了能级下降的数据,而且通过Δρ图真切地看到了电子向中间移动、形成双方电子缺失,使原子成键后形成的能级大大下降的实体形象、具体的物理机制。
2.2. 氢分子二聚体的Δρ
考虑氢分子二聚体相互之间的范德华作用,首先还是要比较二者组成范德华体系时的能量变化情况,考察在范德华作用距离时能量的低谷,其次作电子密度差探索这个低谷的形成机制。
氢分子二聚体的计算研究历来有许多工作[4-7]。二聚体中氢分子的相互组成方式有:相互垂直式T、相互交叉式X、平行有一定位错式Z、相互平行式H和头尾相连式L。
对上述各式分别进行计算优化,二氢分子在各式中的作用距离与对应的能量依次如下:
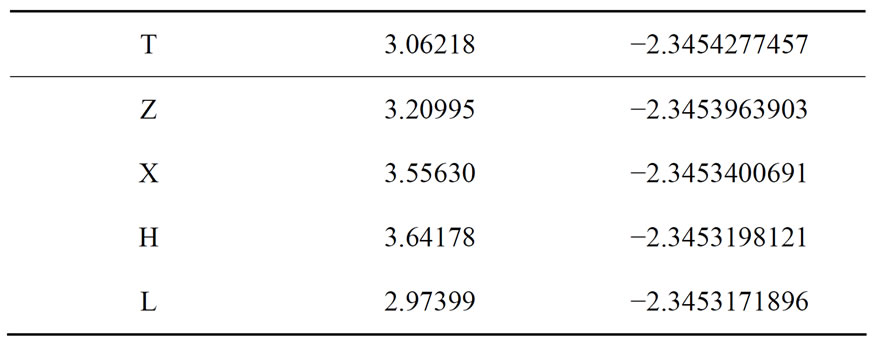
本文没有对各式进行作用距离和能量的更进一步的精确计算,也没有对这些组合方式按能量高低作出选择,只是专门考察这些方式形成范德华作用时的具体机制,因为从相应的电子密度差来考察范德华作用的机制看,已经足够了。
下面以头尾相连式L、相互垂直式T和相互平行式H为例,分别进行考察。
2.2.1. 头尾相连式L
前文[1]表明,在Rg二聚体形成时,因Rg已是配对电子,在二Rg相遇、相碰时,在Rg之间需要挤走部分电子才形成范德华作用。
现在的氢分子,它也是配对电子,但是从图1可以看到,由于电子往键中间聚集,两端的氢已经减少了较多电子,此处电子对核屏蔽减弱,电负性增强,已经为头尾相连形成范德华作用提供了条件。
计算头尾相连式二聚体结果数据如下:
二体中氢分子键长:0.742928。
二体中范德华作用距离:2.973993。
自由氢分子能量:−1.1726355840。
二体总能量:−2.3453171896。
这里,范德华作用距离指二氢分子距离。
氢分子二聚体的电子云图,是两个微椭的圆球中间稍有粘连。
为了观察二聚体电子变化,还是作二聚体与二自由氢分子的电子密度差。见图2。图2的上部是等值面图,等值面的值为±0.00001。紫色增加区域的电荷量记为+0.002638;青兰色减少区域的电荷量记为−0.002635。从这个电荷变化量看,它比形成氢分子的电荷变化量要小约两个数量级。两边减少的量正是中间增加的量,与图1氢分子何其相似。
图2的下部是截面图,等值线初始值±0.00001。紫色是电子密度增加区域;暗兰色虚线是电子密度减少区域。
如果用RHF/aug-cc-pVTZ来对氢分子头尾相连二聚体进行优化,氢分子之间拉开至4.7,已经超出该体系的范德华作用的最佳距离了,这是由于RHF没有考虑电子相关,电子排斥计算得过大了,不能正确描述范德华作用。
2.2.2. 相互垂直式T
优化计算氢分子相互垂直式T二聚体结果主要数据如下:
二体中氢分子键长:0.742200(横),0.743200(竖)。
二体中范德华作用距离:3.062182。
自由氢分子能量:−1.1726355840。
二体总能量:−2.3454277457。
这里,范德华作用距离指左氢分子(横)到右氢分子(竖)中心的距离。
作氢分子二聚体与二自由氢分子电子密度差Δρ。见图3。图3的上部是等值面图,等值面的值为±0.00001。紫色增加区域的电荷量记为+0.002907;青兰色减少区域的电荷量记为−0.002909。
图3的下部是截面图,等值线初始值±0.00001。紫色为正,是电子密度增加区域;暗兰色虚线为负,是电子密度减少区域。
2.2.3. 相互平行式H
优化计算氢分子相互平行式H二聚体结果主要数据如下:
二体中氢分子键长:0.742972。
二体中范德华作用距离:3.64178。

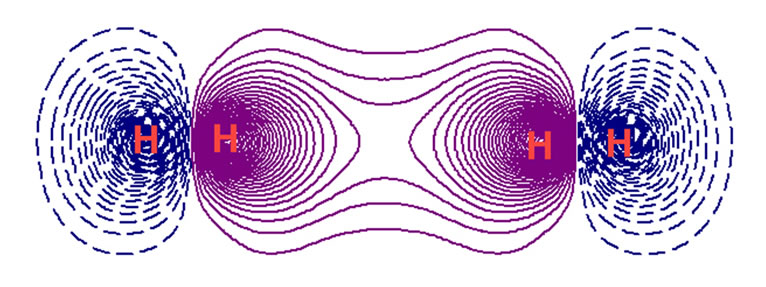
Figure 2. (H2)2(L) electron density differences
图2. (H2)2(L)的电子密度差
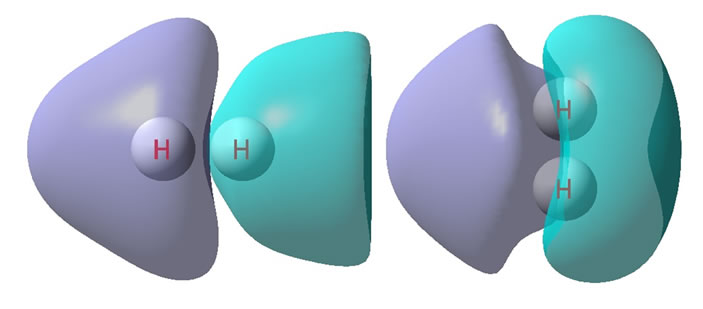

Figure 3. (H2)2(T) electron density differences
图3. (H2)2(T)的电子密度差
自由氢分子能量:−1.1726355856。
二体总能量:−2.3453198121。
这里,范德华作用距离指二氢分子距离。
作相互平行式二聚体与二自由氢分子的电子密度差。见图4。
图4的上部是等值面图,等值面的值为±0.000001。
紫色增加区域的电荷量记为+0.0010933;青兰色减少区域的电荷量记为−0.0010944。
图4的下部是截面图,等值线初始值±0.000001。
紫色为正,是电子密度增加区域;暗兰色虚线为负,是电子密度减少区域。
2.3. 氢分子与氦原子的Δρ
计算氢分子与氦原子体系,优化结果是:氢分子键长0.74282,氢分子与氦距离2.99859。
作氢分子与氦体系与自由氢分子、自由氦的电子密度差。见图5。
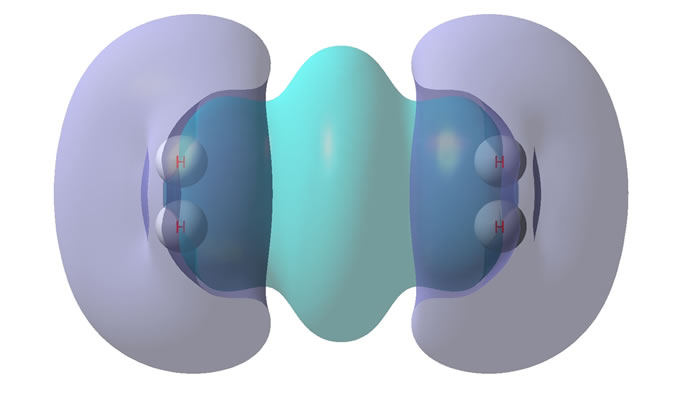

Figure 4. (H2)2(H) electron density differences
图4. (H2)2(H)的电子密度差

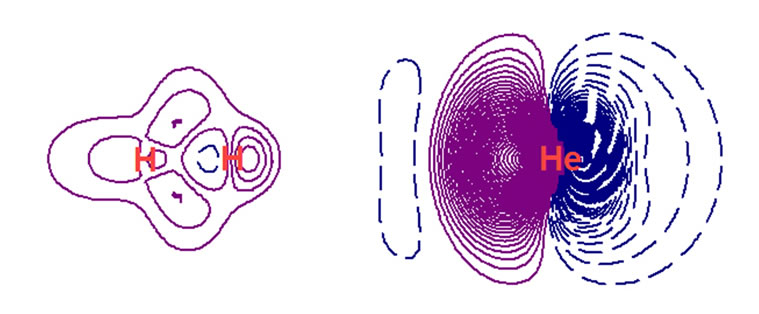
Figure 5. H2He electron density differences
图5. H2He的电子密度差
图5上部是等值面图,等值面的值为±0.0000002。
紫色增加区域的电荷量记为+0.0008304;青兰色减少区域的电荷量记为−0.0008303。
图5的下部是截面图,等值线初始值±0.0000003。紫色是电子密度增加区域;暗兰色虚线是电子密度减少区域。
2.4. 氢分子与氖原子的Δρ
计算氢分子与氦原子体系,优化结果是:氢分子键长0.74285,氢分子与氦距离2.99891。
作氢分子与氦体系与自由氢分子、自由氦的电子密度差。见图6。
图6的上部是等值面图,等值面的值为±0.000002。
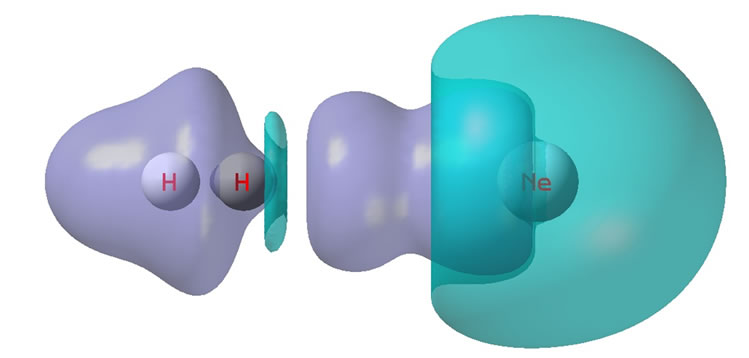

Figure 6. H2Ne electron density differences
图6. H2Ne的电子密度差
紫色增加区域的电荷量记为+0.001275;青兰色减少区域的电荷量记为−0.001272。
图6的下部是截面图,等值线初始值±0.000002。紫色是电子密度增加区域;暗兰色虚线是电子密度减少区域。
3. 图形解析和范德华作用的讨论
1) 前文[1]说稀有气体原子相遇、相碰,双方配对电子在核外形成惰性的电子云球会产生电子的Pauli互斥,如果二者只是刚性的电子云球,它们会即碰即离。但电子云球却不是刚性的,它们相碰,电子云球会产生变形,二者中间的电子会移到二核背面,二核面对之处形成一定程度的裸露,此处吸引电子的能力增强,剩余电子浓缩,从而形成不可忽视的电子共享,即范德华力。这种情形,被电子密度差准确地展示出来了。
对于氢分子,也是配对电子在核外形成电子云球,但从图1可以看到,由于电子往键中间聚集,两端的氢已经减少了较多电子,此处电子对核屏蔽减弱,电负性增强,已经为形成范德华作用提供了基本条件。
于是像图2那样,氢分子头对头地靠近,这里电负性较高,形成势阱,电子会有点往这里聚集,形成了电子共享的增加,形成了范德华作用。
于是像图3那样,氢分子相互垂直地靠近,一方面看到由于在竖向氢分子电子的冲击下,横向的氢分子电子云后退了。横向的氢分子电子云后退的结果,使它朝向竖向氢分子一端电负性增强,竖向氢分子的电子云便朝向横向的氢分子了,增加了共享,形成了范德华作用。
于是像图4那样,氢分子相互平行地靠近,氢分子中部本来就电子密集,互斥很大,产生电子互让,互让使二氢分子之间电子减少,这时突出的是,氢分子端头原本就有所裸露、电负性高,双方相向延伸增加了共享电子。当中间电子互斥较大时,便在边沿增加了共享电子。
电子是如此的灵动,它总是流向能量的低处,使体系能量均衡。
这些,都产生了二氢分子之间的电子共享,但是由于配对电子互斥,电子共享是很有限的,正是在互斥的前提下又发生了共享吸引,达到平衡,形成了范德华作用。这种情形,量子化学CCSD计算已经精确地给出了,同样被电子密度差准确地展示出来了。
纵观分子之间形成范德华作用的全过程,从分子双方相遇、相碰的一开始,伴随着双方电子互斥、互让,电子共享产生的吸引力就同时开始伴生了。而且在初始阶段引力还微大于斥力,导致双方距离不断缩短,斥力和引力都同时在增加。直至引力和斥力达到平衡,就形成了范德华作用最佳距离和作用能。
可见,原子和分子间范德华作用本质和机制是一样的。
当然,如果说得再细一些,这里的斥力除了双方配对电子的Pauli互斥之外,自然还应包含双方核的排斥。以及引力除了共享电子之外,自然还应包含共享电子处在双方的核之间,对双方核的排斥有所屏蔽。只说双方配对电子发生互斥、双方共享电子产生引力,是为了简化而抓主要矛盾。
2) 这里,范德华作用及其机理自然而然地由量子化学定量、可视地描述清楚了,这里并没有“瞬间偶极矩诱导”的假说。设想在超低温和超高压的作用下,氢分子逐渐失去自由活力,以各种形式通过范德华作用相连(特别是能量最低的T式)并多聚延长,直至形成实验测定的6个氢分子成环的晶体,怎样来发生“瞬间偶极矩诱导”?
对于分子间的范德华作用,就是这种分子之间在配对电子排斥的前提下,又产生了一点共享电子的吸引,达到平衡,形成了范德华作用。
“瞬间偶极矩诱导”的假说是基于静电作用的,由此得到的推论是范德华作用在分子间是无方向性、无饱和性的。
然而,在计算氢分子二聚体时,对二者的多种组合方式进行优化,不外乎T式、Z式、X式、H式和L式,T式还最稳定,说明不仅有严格的方向性,而且有严格的饱和性。
其实,仅由范德华作用所形成的许许多多分子晶体,都有严格固定的结构,这些都表明范德华作用是有方向性和饱和性的。
3) 为了使问题简化,前面选择了稀有气体原子二聚体,选择了氢分子二聚体,都是为了回避在二者之间有电子转移的情况,只关注单纯的范德华作用引起的电子净变化。然而现在算的氢分子与氦、氖形成的范德华作用体系,以及相互垂直式氢分子二聚体,已经明显地在形成些许共享电子的同时,有电子转移伴随着了。其实在前面不同稀有气体原子之间的范德华作用二聚体,已经有电子转移了,只是当时注意点只在电子共享,没有把电子转移加以讨论。
这种电荷转移的计算,需要计算电子密度差各区块的电荷量,这只需将得到的电子密度差Δρ的cub文件,读入Multiwfn进行盆分析(basin analysis)就可以得到。
氢分子与氦形成的范德华作用体系,对照图5看,He明显分为左右两半,左半是电子共享的增加量,盆分析对此的电荷量记为+0.0006143,右半是电荷中移共享发生的电荷减少量,盆分析的电荷量记为−0.0006311。然而减少的量比增加的量多出了0.0000186,这部分电荷转移到氢分子上了,氢分子上则多出了这个量。
氢分子与氖形成的范德华作用体系,对照图6看,Ne也明显分为左右两半,左半是电子共享的增加量,盆分析对此的电荷量记为+0.0008803,右半是电荷中移共享发生的电荷减少量,盆分析的电荷量记为−0.001185。然而减少的量比增加的量多出了0.0003047,这部分电荷转移到氢分子上了(氢分子增加量为+0.0003159,它还自筹了一点点)。
至于相互垂直式氢分子二聚体系,对照图3看,左边是是横向的,右边是竖向的。
竖向的氢分子明显分为左右两半,左半是电子共享的增加量,盆分析对此的电荷量记为+0.001587,右半是电荷因向中间移动共享发生的电荷减少量,盆分析的电荷量记为−0.001472。然而左半增加的量比右半减少的量多出了0.000115,这部分电荷是由横向的氢分子转移过来的。
对照图3还可以看到,横向的氢分子也明显分为左右两半,左半电子明显增加了,盆分析对此的电荷量记为+0.001319,右半是电荷相应的减少量,盆分析的电荷量记为−0.001436。横向的氢分子少了0.000117,转移到竖向氢分子了。
当然这些电荷量都很微细,但是范德华作用就是发生在这种微细电荷量变化之上的,范德华作用也是微细的,二者在数量级上是匹配的。
4) 对通常的共价键,可以理解为原子或分子片各出一个电子,自旋方向相反,形成配对电子,电子在原子或分子片之间按电负性大小分配,电负性大的得到电子,电负性下降,电负性小的失去电子,电负性上升,直至二者电负性均衡,通过电子流动调整电负性来达到电负性均衡,此即电负性均衡原理[8]。电负性均衡也即能级均衡。
然而在氢分子与氦、氖形成的范德华作用体系,从元素周期表电负性看,氦(3.89)、氖(3.62),电负性都很高,怎么会电子流向氢分子呢?氦、氖是惰性的,怎么还有如此高的电负性?
用RHF/aug-cc-pVTz算单个的氦、氖得到下面结果。
He的HOMO为−0.91787,与实验测得的第一电离能相符,表明电子不易失去;而LUMO为+0.11354,表明不易得到电子。
Ne的HOMO为−0.85121,与实验测得的第一电离能相符,表明电子不易失去;而LUMO为+0.24081,表明不易得到电子。
氦、氖就是这样既不易失去电子,也不易得到电子。所以才称为惰性气体。
然而CCSD可信的精确计算和Multiwfn进行盆分析得到的电荷,表明在氢分子与氦、氖形成的范德华作用体系中,确实有些微的电荷流向氢分子了。
看来,范德华作用体系与共价键双方各出一个电子一拍即合形成电子共享还有所不同。
范德华作用体系最基本特征是双方各自都已是配对电子。当它们相遇、相碰时,是从双方配对电子的强大的Pauli互斥开始的。
范德华作用体系是在配对电子互斥、制约之下形成的些微的电子共享。
对氢分子与氦、氖形成的范德华作用体系可以这样设想,氦、氖由于吸引自身电子的能力比氢分子强,氢分子与氦、氖相遇、相碰时,相比之下较为刚性一些的氦、氖对氢分子产生冲击,使氢分子电子分布发生变形,相比之下氢分子更有电子退让的余地,这种变形在图5、6展示得很明显。
在图1所展示的氢分子Δρ中,本来H的外端已经减少电子了,在氦、氖电子的冲击下,H的这个外端电子更减少了,电负性更增强了,它对氦、氖电子吸引力增强了,在图5、6,氦、氖的电子云明显趋向于氢分子。氦、氖朝向氢分子的这一面电子明显增多,又使得氦、氖的这一面电负性下降,终于发生了逆转,氦、氖在此处的电负性反倒比端氢稍低,氦、氖的些微电子要流向氢分子,以达到电负性均衡,图5、6正是电负性均衡后的状况。
至于相互垂直式氢分子二聚体系,之所以发生电子云演变,是由于在双方配对电子的强大的Pauli互斥下,横向的氢分子比起竖向氢分子,较有纵深、更易于变形一些,在竖向氢分子的冲击下,横向的氢分子电子云退让了。横向的氢分子电子云后退的结果,使它朝向竖向氢分子一端电负性增强,竖向氢分子的电子云便朝向横向的氢分子了,增加了共享。但总体来看,对共享电子而言,竖向氢分子有两个核吸引它,有微量的电子转移到竖向氢分子上了,从而达到电负性均衡,图3正是电负性均衡后的结果。
电子在范德华体系的分子间也有转移,这是它们在分子中电负性发生变化所驱动的,并由于电子在分子间转移使范德华作用的两边达到电负性均衡。
4. 结论
范德华作用是在分子双方配对电子互斥、制约之下形成的些微的电子共享。是配对电子互斥与共享电子吸引的平衡。
范德华作用是作用双方电子轨道相互作用的结果,属于化学作用。范德华作用是有方向性和饱和性的。
分子双方配对电子的相互作用,使分子双方在作用体系中的实际电负性发生了变化,依据这种变化了的实际电负性差距,引起电子流动,从而达到电负性均衡,遵循电负性均衡原理。
从共享电子产生分子间引力,电子流动并达到电负性均衡,有方向性、饱和性看,范德华作用与通常的共价概念在内容和形式上都是有共通之处的。
5. 致谢
Sobereva博士对量子化学计算和文献给予了指导,对此研究工作进行了讨论,特此致谢!
参考文献 (References)
[1] 周光耀 (2013) 范德华作用的量子化学研究(一). 物理化学进展, 2, 21-26.
[2] The Gordon Research Group (2007) The General atomic and molecular electronic structure system (GAMESS) is a general ab initio quantum chemistry package.
[3] Lu, T. and Chen, F.W. J. (2012) Multiwfn: A multifunctional wavefunction analyzer. Computers & Chemistry, 33, 580-592.
[4] van Kranendonk, J. and Gush, H.P. (1962) The crystal structure of solid hydrogen. Physics Letters A, 1, 22.
[5] Ree, F.H. and Bender, C.F. (1979) Repulsive intermolecular potential between two H2 molecules. Journal of Chemical Physics, 71, 5362-5375.
[6] Schneider, B., Hbza, P. and Zahradnik, R. (1988) Potential energy surface of the (H2)2 dimer: An MP2 study. Theoretical Chemistry Accounts, 73, 201-206.
[7] Kim, C., Kim, S.J., Lee, Y. and Kim, T.Y. (2000) Quantum mechanical study of van der waals complex, I. The H2 dimer using the DFT and the multi-coefficient G2/G3 methods. Bulletin of the Korean Chemical Society, 21, 510-514.
[8] 周光耀 (1985) 关于电负性均衡原理. 化学学报, 1, 1-3.