Journal of Advances in Physical Chemistry
Vol.04 No.04(2015), Article ID:16370,14 pages
10.12677/JAPC.2015.44013
Generalized Charge Decomposition Analysis (GCDA) Method
Meng Xiao1, Tian Lu2*
1Patent Examination Cooperation Center of the Patent Office, SIPO, Beijing
*通讯作者。
2Beijing Kein Research Center for Natural Sciences (www.keinsci.com), Beijing
Received: Oct. 28th, 2015; accepted: Nov. 11th, 2015; published: Nov. 19th, 2015
Copyright © 2015 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



ABSTRACT
Charge decomposition analysis (CDA) is an important method to study charge transfer problems in depths. In this paper, we introduced CDA method, elucidated the issues that needed attention in the calculation, and presented a new form of CDA, which was called GCDA. At HF and DFT levels, GCDA is equivalent to CDA, while GCDA is more applicable at post-HF level, and meantime its open-shell form is explicitly considered. By taking OC-BH3 and cis-dichlorodiamine as test cases, we find the GCDA results at various theoretical levels are reasonable and qualitatively consistent, but differ quantitatively; in particular, a rough trend exists in the d term: Functional with low HF composition > Functional with high HF composition > HF ³ MP2 > CCSD. The paper also discussed the choice of basis-set in GCDA calculation.
Keywords:Charge Decomposition Analysis, Charge Transfer, Quantum Chemistry

广义化的电荷分解分析(GCDA)方法
肖萌1,卢天2*
1国家知识产权局专利局专利审查协作北京中心,北京
2北京科音自然科学研究中心(www.keinsci.com),北京

收稿日期:2015年10月28日;录用日期:2015年11月11日;发布日期:2015年11月19日

摘 要
电荷分解分析(CDA)是一种重要的深入研究电荷转移问题的方法。本文对CDA进行了介绍,阐述了计算中需要注意的问题,并提出了一种新的CDA形式,称为广义化的电荷分解分析(GCDA)。在HF、DFT级别GCDA与CDA等价,但GCDA能更好地用于后HF计算,而且还明确考虑了开壳层时的计算形式。通过以OC-BH3和顺式二氯二胺合铂体系作为测试实例,我们发现各种理论级别下GCDA的结果都较为合理且定性相符,但在定量上有所区别,特别是d项存在大体趋势:低HF成份的泛函 > 高HF成份的泛函 > HF ³ MP2 > CCSD。文中还对GCDA计算时基组的选择进行了讨论。
关键词 :电荷分解分析,电荷转移,量子化学

1. 引言
两个或多个分子片段间形成化学键时总是伴随着电荷在片段间的转移。对电荷转移的分析考察是量子化学理论研究中的重要组成部分。考察电荷转移的常见方式有两种:1) 作密度差图[1] ,即令整个体系的电子密度减去处于相同几何结构下的两个片段的电子密度,这可以十分直观地考察电子在不同区域的增、减,但不便于定量考察;2) 对整个体系计算原子电荷[2] ,再对每个片段在其孤立状态下计算原子电荷,比较结果的差异,可以确切得知成键前后各个原子的电子得、失量。若将实际体系每个片段内的原子电荷加和成为片段电荷与参考状态相对比,还可以得知成键是如何影响整个片段所带电荷的。另外,如果用Mulliken、自然布居分析(NPA)等方法[2] -[4] 做布居分析,还可以考察成键前后各原子轨道所含电子数的变化。
然而,上述两类分析方式只能得到电荷转移的总结果,更深入、细节的信息无法得到。例如,当体系由多个片段构成,没法考察电荷在每一对片段间的转移量。再比如研究过渡金属配合物的配位键时往往想要考察配体向金属贡献的电子量,以及金属向配体反馈的电子量,但上述分析没法得到这样的信息,而只能得到净转移量。1995年S. Dapprich和G. Frenking提出了电荷分解分析(Charge decomposition analysis, CDA)方法[5] ,可以对电荷转移问题做更透彻的分析,能够解决以上问题。CDA方法已被应用于大量问题的研究[5] -[16] ,成为电荷转移问题的重要分析手段。
2. CDA方法
在CDA方法中,整个体系被称复合物。复合物的轨道被视为由各个片段的片段轨道线性组合而成,片段轨道数目总和与复合物轨道数目一致。复合物轨道是在优化的复合物结构下做单点能计算得到的,片段轨道是对各个片段分别做单点能计算得到的,所用的片段结构直接从优化的复合物结构中提取。CDA方法定义了三个量,d (Donation)表现的是片段A向片段B贡献的电子量,b (Back-donation)表现的是片段B向片段A反馈的电子量,r (Charge polarization)表现的是占据轨道的电荷极化。在CDA方法中这三个量可以对组成体系的任意两个片段计算,而且可以分解为各个复合物轨道独立的贡献。第i复合物轨道对片段A、B间的d, b, r项的贡献由下式计算
 (1)
(1)
其中m代表A片段的片段轨道,n代表B片段的片段轨道,上标occ和vir分别代表加和只循环占据和非占据片段轨道,hi代表i复合物轨道的占据数。Cm,i代表i复合物中m号片段轨道的展开系数,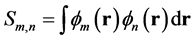 代表m和n片段轨道间的重叠积分。
代表m和n片段轨道间的重叠积分。
(1)式的di表现了经由第i复合物轨道片段A向片段B转移的电子量,由表达式可见它体现的是片段A的占据轨道与片段B的空轨道的混合。bi表现了经由第i复合物轨道片段B向片段A反馈的电子量,它由片段B的占据轨道与片段A的空轨道混合而产生。di-bi可以认为是经由第i复合物轨道两个片段间的净转移量。若令di、bi、di-bi对所有复合物轨道进行加和得到d、b、d-b,就能得知A对B的总贡献量、B向A的总反馈量,以及净转移量。另外,还可以定义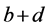 ,其数值越大说明两个片段间相互作用程度越大,轨道混合越显著。由(1)式可见r项是两个片段占据轨道间的作用,CDA方法中对它物理意义的解释是:若ri
> 0,说明由于第i个复合物轨道,两个片段的电子在片段重叠区域的分布量增加,此时两个片段的占据轨道以相位相同方式混合,起到成键效应;若ri
< 0,表明两个片段的电子在重叠区域分布量减少,此时两个片段的占据轨道间必以相位相反方式混合,对片段间成键起到破坏作用,即反键效应。对所有复合物轨道的ri的总和值r一般为负值,因为占据轨道间的总相互作用是以交换互斥为主导的,是不利于成键的。由于r不涉及到电荷转移问题,而只体现出因占据片段轨道的相互作用导致电子密度分布发生极化,因而称为电荷极化项。
,其数值越大说明两个片段间相互作用程度越大,轨道混合越显著。由(1)式可见r项是两个片段占据轨道间的作用,CDA方法中对它物理意义的解释是:若ri
> 0,说明由于第i个复合物轨道,两个片段的电子在片段重叠区域的分布量增加,此时两个片段的占据轨道以相位相同方式混合,起到成键效应;若ri
< 0,表明两个片段的电子在重叠区域分布量减少,此时两个片段的占据轨道间必以相位相反方式混合,对片段间成键起到破坏作用,即反键效应。对所有复合物轨道的ri的总和值r一般为负值,因为占据轨道间的总相互作用是以交换互斥为主导的,是不利于成键的。由于r不涉及到电荷转移问题,而只体现出因占据片段轨道的相互作用导致电子密度分布发生极化,因而称为电荷极化项。
需要注意的是,d和b项除了表现出电子的转移外,实际上也同时混杂着电子极化效应的贡献。因为当A片段的占据轨道与B片段的空轨道混合时,实际上不光是A的电子向B转移,B的空轨道还使得A的电子在A自身区域分布被极化。当极化程度很大时,d、b可能分别和实际的电子贡献量、反馈量有明显偏差。因此原理上d-b并不能准确衡量电荷净转移量,而只适合用于定性考察。对于只含两个片段的情况,若只需要考察片段间电子净转移量,用合适的方法计算原子电荷并加和为片段电荷来分析更为可靠。扩展的CDA方法(Extended CDA, ECDA)方法[6] 则在CDA基础上专门将极化和电荷转移效应进行分离,从而能较可靠地获得电子净转移量,但ECDA方法只能用在包含两个片段的情况,而且无法将d、b分离给出,因此实用性很有限,本文也不做更多讨论。
3. 对CDA方法的广义化
原始的CDA方法可以基于Hartree-Fock (HF)或密度泛函理论(DFT)得到的分子轨道进行计算,但是用于后HF方法则有一定局限性。CDA公式中有复合物轨道的占据数一项,因此CDA原文中认为如果使用后HF计算复合物时得到的自然轨道的占据数,而片段轨道用HF方法来计算,则CDA也可以用在后HF波函数上,但我们认为这种做法所不妥。因为在后HF级别下做CDA分析,只有复合物和各个片段都使用后HF方法的自然轨道才能保持整体计算级别的一致性。然而,原本的CDA方法中并没有考虑到片段轨道的占据数,而仅仅区分为占据和非占据。为解决此问题,从而使得CDA分析在后HF级别下更为合理,本文我们提出一种CDA方法的新形式,称广义化的电荷分解分析(Generlized CDA, GCDA),它将di和bi合并为统一的表达式ti:
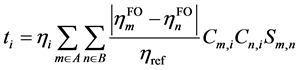 (2)
(2)
相比(1),这里额外引入了片段轨道的占据数hFO,以及参考占据数href (体现轨道占据数的理论最大值),并且加和范围涉及全部片段轨道,不再区分占据和非占据。di项的计算方法是在计算ti时只对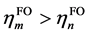 的情况进行加和,计算bi时只对
的情况进行加和,计算bi时只对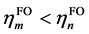 的情况进行加和。由于GCDA将片段轨道的占据数直接考虑了进去,故片段轨道像复合物轨道一样可以是分子轨道也可以是后HF方法得到的自然轨道。GCDA计算时复合物和片段计算的方法应一致,如果用后HF方法产生复合物的自然轨道,则片段也应当用同样级别产生片段自然轨道;如果复合物用的是分子轨道,则片段也应当用相同级别产生片段分子轨道。
的情况进行加和。由于GCDA将片段轨道的占据数直接考虑了进去,故片段轨道像复合物轨道一样可以是分子轨道也可以是后HF方法得到的自然轨道。GCDA计算时复合物和片段计算的方法应一致,如果用后HF方法产生复合物的自然轨道,则片段也应当用同样级别产生片段自然轨道;如果复合物用的是分子轨道,则片段也应当用相同级别产生片段分子轨道。
GCDA的定义有清晰的物理意义。从(2)式可见,如果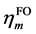 和
和 的占据数都一样,则二者彼此间不会有电子转移。二者占据数差值越大,则转移程度越大,而且由占据数高的一方流向低的一方。若其中一方被整数占据而另一方占据数为0,表达式就会自动恢复为原始CDA的定义,即(1)式。
的占据数都一样,则二者彼此间不会有电子转移。二者占据数差值越大,则转移程度越大,而且由占据数高的一方流向低的一方。若其中一方被整数占据而另一方占据数为0,表达式就会自动恢复为原始CDA的定义,即(1)式。
原始的CDA方法并未明确考虑如何用于开壳层体系,而GCDA明确考虑了这一点。在GCDA计算中,若复合物和片段均为闭壳层,则对它们做限制性计算,用得到的分子轨道或自然轨道做闭壳层形式的GCDA,此时href = 2.0。反之,若有任何一个片段为开壳层,则应当对所有体系做非限制性计算得到a和b自旋分子轨道或自旋自然轨道(对其中闭壳层体系为省时也可以做限制性计算但之后再把无自旋轨道等分为a和b自旋轨道),然后以开壳层形式做GCDA,此时href = 1.0。开壳层形式的GCDA中a和b自旋是单独处理的。例如,计算a部分时,(2)式的i, m, n循环的是a分子轨道(HF或DFT计算时)或者a自旋自然轨道(非限制性后HF计算时)。开壳层形式的GCDA在分析时需要单独分析a和b两种自旋的结果,整体的d, b, r项是a和b相应项的加和。
GCDA对r项的定义也与CDA不同,为
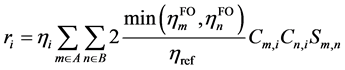 (3)
(3)
其中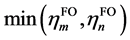 代表取
代表取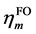 和
和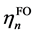 其中的较小值,表现出两个片段轨道间的互斥作用由二者共有的电子数来决定。因此若有一方占据数较小则ri必然也较小,仅对于两个高占据轨道间ri项可能很大。对于原先CDA适用的情况,即片段轨道占据数为整数时,GCDA得到的ri是原先CDA定义的ri的二倍。之所以相比CDA额外引入了因子2,是因为这样定义时ri里的每一项会类似于m和n在i中的重叠布居,这样物理意义更明确。在Mulliken布居分析[4]
[17] 中,a, b两个基函数在i轨道中的重叠布居由下式定义,可以与(3)式相对照
其中的较小值,表现出两个片段轨道间的互斥作用由二者共有的电子数来决定。因此若有一方占据数较小则ri必然也较小,仅对于两个高占据轨道间ri项可能很大。对于原先CDA适用的情况,即片段轨道占据数为整数时,GCDA得到的ri是原先CDA定义的ri的二倍。之所以相比CDA额外引入了因子2,是因为这样定义时ri里的每一项会类似于m和n在i中的重叠布居,这样物理意义更明确。在Mulliken布居分析[4]
[17] 中,a, b两个基函数在i轨道中的重叠布居由下式定义,可以与(3)式相对照
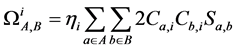 (4)
(4)
4. CDA/GCDA方法的计算要点
CDA或GCDA的实际计算中有一些要点和常见问题我们认为很值得强调:
1) 使用CDA方法进行计算时不需要很大基组,通常用6-31G** [18] 、cc-pVDZ [19] 、def2-SVP [20] 等双zeta基组足矣得到合理结果。含有弥散函数的基组应当避免使用,因为弥散函数并没有明确的化学意义,在计算片段轨道时弥散函数往往会使得非占据轨道表现出弥散特征,这会使得CDA/GCDA方法的计算形式很大程度失去物理意义。例如,如果片段B的空轨道弥散范围极大,严重侵入片段A的空间中,那么当计算A®B的d项时,这个值将主要体现B的空轨道与A的占据轨道的混合使得A区域的电子密度发生极化,而不是主要体现A的占据轨道的电子向B片段所在区域的转移。基组问题我们将在后文通过实例进行专门讨论。
2) 各个轨道对d、b值的贡献大多数情况为正值,表现电子贡献和反馈量,但也有少数情况为负值。较小的负值在讨论时可以忽略不计,也并没有确切的物理解释。当出现较大的负值时,说明计算过程可能存在问题,或者CDA/GCDA并不完全适用于当前体系。我们也注意到,当使用弥散函数时,总是倾向于引起大量负值的出现,一方面表明弥散函数会导致结果不合理,也同时从侧面说明一些轨道对d、b的负贡献很大程度上和片段内电子密度的极化有关。
3) CDA/GCDA计算要求片段轨道数总和必须等于复合物轨道数目,这首先要求计算片段时对各个原子所用的基组必须与它们在复合物计算时所用的基组严格相对应,因为计算得到的分子轨道数与基函数相等。这不仅要求基组名相对应,还需要注意基函数类型也应当一致,因为对d、f、g壳层,球谐型高斯函数分别有5、7、9个基函数,而笛卡尔型高斯函数则分别有6、10、15个基函数。一个经常需要注意的情形是,用主流的Gaussian09程序计算过渡金属配合物,对配体原子用6-31G*基组,对过渡金属用Lanl2DZ [21] 赝势基组,此时程序在复合物计算时会默认使用球谐型高斯函数,然而当单独用6-31G*计算配体片段时,程序会默认用笛卡尔型高斯函数,这样就导致计算复合物时的基函数数目(复合物轨道数)与所有片段的基函数数目之和(片段轨道数之和)不一致,从而无法进行CDA/GCDA计算。因此,计算时应当令量子化学程序要么都用笛卡尔型高斯基函数,要么都用球谐型高斯基函数。另外,还应注意很多量子化学程序会自动去除线性依赖基函数以保证数值稳定性,这会使得算出的轨道数少于基函数数目,同样会造成复合物和片段轨道数不一致的问题。线性依赖问题通常只在使用了弥散函数的情况下出现,因此从这个角度上也应避免使用弥散函数。
4) 虽然原理上CDA/GCDA并不严格要求计算片段时所用的几何结构必须与它在复合物结构中一致,但如果不满足这个要求结果很可能不合理。因此,实际计算时,应当先优化复合物,然后直接取出其中片段的坐标,再对片段做单点能计算得到片段轨道。同时应注意一些量子化学程序为了利用对称性,会自动调整输入文件里的坐标。例如Gaussian09程序在计算前会自动将体系摆到标准朝向下,这显然会导致复合物坐标与片段坐标出现不对应性,利用nosymm关键词则可以避免程序自动调整朝向。
5) 以开壳层方式做CDA/GCDA计算时需要考虑自旋翻转问题。量子化学计算时自旋多重度等于体系a电子数减b电子数加1,因此自旋多重度 > 1的体系在计算时都是a电子数多于b电子。CDA/GCDA计算时要求片段的a电子数之和等于复合物a电子数,片段的b电子数之和等于复合物b电子数。但实际情况中,通常需要经过自旋翻转才能满足这个条件。例如计算CH3Cl,甲基和氯原子各作为一个片段,整个体系有13个a电子和13个b电子。计算甲基自由基时有5个a电子和4个b电子,计算氯原子时有9个a电子和8个b电子,直接加和为14个a电子和12个b电子,显然和整体不符。因此在做CDA/GCDA时,必须将Cl的a和b轨道交换,相应地a电子数和b电子数也进行交换,这样片段的电子数和电子结构才与复合物匹配。
6) 做CDA/GCDA分析时在片段的参考状态的选择上有一定任意性。例如KCl体系,单独计算片段时既可以让K和Cl的电荷都为0,相当于以中性自由基为片段状态;也可以让二者电荷分别为+1和−1,相当于以离子为片段状态。显然两种参考状态的分析结果会显著不同。参考状态的选择,应当尽量接近复合物状态的电子结构。因为KCl的电子结构可近似写为K+Cl−,因此以离子为参考状态更为合理,分析结果将主要表现的是氯阴离子如何转移电子给钾阳离子。
7) 哪个片段作为给体和受体片段并不影响结果。例如将KCl中的Cl−设为给体片段,K+设为受体片段,则d项很大而b项甚微;如果将K+设为给体片段,Cl−设为受体片段,则b项很大而d项甚微。因此交换给体、受体片段,只是令d、b项交换,r项则不会变化。但为了方便讨论,建议将主要体现给电子的片段作为给体片段,从而使d-b为正值。
5. 分析实例
下面我们将通过两个简单实例,即OC-BH3和顺式二氯二胺合铂来检验GCDA方法的结果,这里我们的目的不在于充分展现GCDA的应用价值,而侧重于考察GCDA定义的合理性,以及计算条件对结果的影响。几何优化我们采用B3LYP/def2-TZVP [20] [22] 级别,这样级别下优化的几何结构对于当前测试体系是很可靠的。若未注明,在CDA/GCDA分析时所用基组为def2-SVP [20] 。几何优化和波函数的产生使用ORCA 3.0.3程序[23] 。CDA/GCDA计算使用我们之一卢天开发的多功能波函数分析程序Multiwfn 3.3.8版[24] [25] ,它能够支持Gaussian、ORCA、Molpro等主流量子化学程序,可以定义无数个片段,可以输出每一对片段轨道对每个复合物轨道di、bi、ri项的贡献以便于考察片段轨道的混合,能够输出片段在复合物轨道中所占成分,还支持前面提到的ECDA方法。本文的轨道图形也通过Multiwfn程序绘制,原子偶极矩校正的Hirshfeld (ADCH)原子电荷[26] 也由Multiwfn计算。HF和DFT计算时CDA与GCDA等价,下文统一称为GCDA,仅在后HF级别计算时才对CDA和GCDA作区分。
5.1. OC-BH3
我们首先以OC-BH3体系作为例子,将CO定义为片段1,BH3定义为片段2来做GCDA分析。我们将考察理论方法、基组对结果的影响,并且分析后HF级别下GCDA和CDA结果的差异。此体系是CO的碳提供孤对电子与BH3形成配位键,因此可以预期电子会从CO明显向BH3转移。我们在HF/def2-SVP下计算了ADCH原子电荷并加和为片段电荷,得到转移量是0.307,通过ECDA方法得到的转移量是0.318。
在HF下和在最常用的DFT泛函B3LYP下[22] 的GCDA详细分析结果列于表1。从表1可见,HF和B3LYP下的GCDA结果都表现出对d项贡献最大的是9号复合物轨道,其次是5号复合物轨道。它们对d-b值的贡献也明显超越其它复合物轨道,因此可以认为CO主要是经由这两条复合物轨道向BH3贡献电子的。从表中也可见对b项贡献最大的是10和11号复合物轨道,因此BH3主要是通过这两条复合物轨道向CO反馈电子的。这两条轨道由于体系对称性而简并,所以反馈量相同。HF和B3LYP下给出的d-b值相当一致,约为0.19,这比起ADCH原子电荷和ECDA方法算出来的转移量要小,这属于正常情况,因为如前文所述,GCDA方法的d、b项还隐含体现了极化效应。HF与B3LYP方法做GCDA给出的结论虽然相符,但不同轨道对d、b、r的贡献量,以及总的d、b、r在定量上有一定差异。B3LYP下的d、b值略大于HF下的,体现出B3LYP下占据片段轨道和非占据片段轨道的混合程度比HF时略大。
如前所述,对于HF、DFT这样轨道为整数占据的情况,CDA和GCDA方法是等价的,但在后HF级别下,由于自然轨道占据数不为整数,二者结果不再一致。做GCDA分析时,对复合物和片段皆使用后HF方法产生的自然轨道,而做CDA分析时,其原文的做法是仅对复合物用后HF自然轨道,而对片段依然用HF轨道。为比较两种做法的差异,表2、表3分别给出了后HF方法中最廉价的二阶微扰理论(MP2)级别和较好质量的单双激发耦合簇(CCSD)级别下做GCDA分析和CDA分析得到的结果。可以看到无论是MP2还是CCSD级别,GCDA和CDA的结果之间无论是在轨道独立贡献上,还是在总值上都很相近。但当前OC-BH3体系的电子结构较为简单,对于电子结构相对复杂一些的二氯二胺合铂配合物是否GCDA和CDA的结果依然差异很小,我们将在下一节进行检验。
值得一提的是,自然轨道没有确切的轨道能量的概念,表2、表3中的复合物自然轨道是按照占据由高到底排序的,为简明我们只列出了前15条。只考虑这15条轨道对d、b、r值的贡献时结果和考虑全部轨道时很接近。另外,12~15号轨道的占据数很低,相当于“准空轨道”,它们对d、b、r的贡献都明显在0.01以下。因此,后HF级别下做GCDA分析时可以忽略那些占据数接近0的复合物自然轨道,除非电子静态相关很强而导致低占据的自然轨道的占据数不很接近于0 (如>0.15)。

Table 1. GCDA results for OC-BH3 system based on HF orbitals and B3LYP orbitals. Significant d, b, r terms are bolded for easier comparison
表1. 基于HF轨道和B3LYP轨道对OC-BH3体系做GCDA分析得到的结果。数值较大的d、b、r以粗体标出以便比较
Table 2. CDA and GCDA results for OC-BH3 system at MP2 level. Significant d, b, r terms are bolded for easier comparison
表2. OC-BH3体系在MP2级别下的GCDA和CDA分析结果。数值较大的d、b、r以粗体标出以便比较
a做GCDA分析时对复合物和片段都用MP2自然轨道。b原始CDA方法在MP2级别的结果,即对复合物用MP2自然轨道,但对片段用HF轨道。为了便于与GCDA的r项对照,r项乘以了2。
Table 3. CDA and GCDA results for OC-BH3 system at CCSD level. Significant d, b, r terms are bolded for easier comparison
表3. OC-BH3体系在CCSD级别下的GCDA和CDA分析结果。数值较大的d、b、r以粗体标出以便比较
a做GCDA分析时对复合物和片段都用CCSD自然轨道。b原始CDA方法在CCSD级别的结果,即对复合物用CCSD自然轨道,但对片段用HF轨道。为了便于与GCDA的r项对照,r项乘以了2。
将表2、表3的轨道贡献数据与表1进行对比,会发现对于d、b、r的贡献值,MP2自然轨道、CCSD自然轨道,以及HF和B3LYP分子轨道之间相差较大,尽管总的d、b、r值相差程度较小。例如,HF和B3LYP的第9条分子轨道对d贡献分别为0.182和0.204,在GCDA方法下MP2和CCSD的第9条自然轨道对d的贡献分别为0.238和0.286。我们还同时注意到,对于当前体系,自然轨道的描述更为紧凑。例如,HF的第5、9号复合物分子轨道对d的贡献分别占总值的32.4%和57.8%,B3LYP时的情况也类似,但是MP2和CCSD下仅9号复合物自然轨道对d的贡献就分别达到了76.8%和96.6%。之所以自然轨道与分子轨道产生的独立贡献相差相当大,是因为两类轨道本身并无严格对应关系,一些轨道的形状也存在一定差异。为了考察清楚这一点,我们在表4当中列出了HF的第9条复合物分子轨道以及CCSD的第9条复合物自然轨道对d、b、r项的贡献细节,并在图1中将涉及到的轨道图形进行了展示,片段轨道对复合物轨道的贡献百分比也标于图中。我们首先考察HF的情况。从表4中可以看到在HF级别下,CO的7号占据分子轨道(主要体现C的孤对电子)与BH3的2号占据轨道(B的2s原子轨道与三个氢的成键轨道)的混合对r的贡献高达−0.771,对照图1可以看到二者明显以相位相反方式重叠,这势必导致两个片段间电子密度显著减小。9号复合物分子轨道的特征实际上也正由这两条片段轨道的混合所主导,二者构成了它的18% + 61.7% = 79.8%的成份。CO的7号占据轨道也同时与BH3的5号空轨道发生了显著混合,这造成了C的孤对电子向BH3转移,对d产生了0.166的贡献。下面我们再来考察CCSD
Table 4. Contribution of the 9th complex HF molecular orbital and the 9th complex CCSD natural orbital to d, b, r terms of GCDA result of OC-BH3 systema
表4. HF下的第9条复合物分子轨道以及CCSD下的第9号复合物自然轨道对OC-BH3体系的GCDA的d、b、r项的贡献a
a为简明起见,仅列出了对d、b、r其中之一贡献大于0.05的片段轨道对。括号中为轨道占据数。
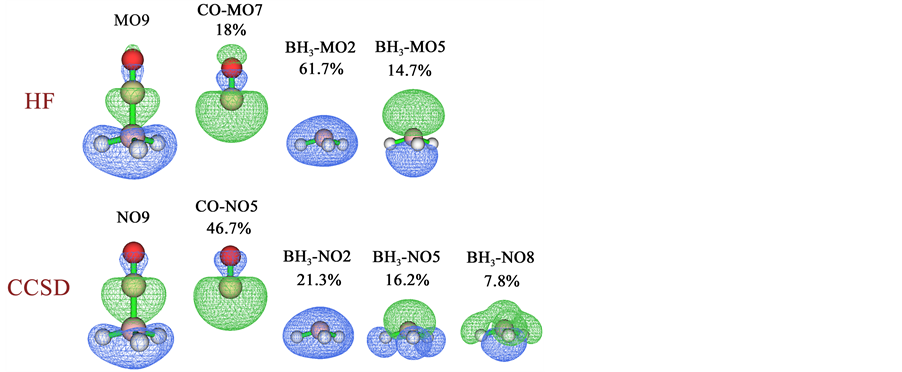
Figure 1. The 9th complex molecular orbital (MO) and some fragment molecular orbitals calculated at HF level, and the 9th complex natural orbital (NO) and some fragment natural orbitals calculated at CCSD level for OC-BH3. The contribution percentages of each fragment orbital to complex orbital derived from Mulliken method are also labelled. The orbital isovalue is set to 0.05, green and blue parts correspond to positive and negative regions of orbital phase, respectively. For ease of comparison, orbital phase is inverted for the fragment orbitals with negative combination coefficient
图1. 对于OC-BH3在HF级别下计算的第9号复合物分子轨道(MO)和部分片段分子轨道,以及CCSD级别下计算的第9号复合物自然轨道(NO)和部分片段自然轨道。Mulliken方法计算的各个片段轨道对复合物轨道贡献的百分比也标于图中。轨道等值面数值为0.05,绿色和蓝色分别代表相位为正和为负的部分。为了便于对比,我们对组合系数为负的片段轨道的相位进行了反转
的情况。结合表4和图1可见CO的高占据的5号片段自然轨道与BH3的2号高占据自然轨道以相位相反方式发生了明显混合,和HF的情况一样对r产生了很大的负贡献(−0.686)。另外,CO的5号片段自然轨道与BH3的两条低占据自然轨道(5号、8号)都以相位相同方式发生了强烈混合,分别导致对d贡献了0.162和0.081。虽然二者之和0.243显著大于HF的0.166,但是是完全合理的,因为其数值从轨道图形上能够很合理地解释。
DFT泛函对GCDA分析结果的影响是值得探究的。我们选择了一些常见泛函,将结果列于表5。考虑的泛函遍及了HF交换成份为0%到100%的情况,包括BLYP [27] [28] 、PBE [29] 、B3LYP、PBE0 [30] 、BH&HLYP [31] 、M06-2X [32] 和M06-HF [33] ,另外还考虑了近程和远程HF成分不同的wB97XD [34] 泛函。从表中数据可见GCDA的结果是明显依赖于泛函的,而且所有泛函都是d、b项大于HF而r项的绝对值小于HF。d-b项受泛函的选择影响则相对较小,基本与HF的值相仿佛。我们也同时发现一个粗略的趋势,即HF成份越大,结果越倾向于接近HF,特别是具有100% HF成份的M06-HF泛函与HF的结果非常相似。wB97XD虽然远程为100%的HF成份,但CDA结果则主要取决于它的近程HF成份(22.2%),其结果与HF成份为25%的PBE0很接近。
表6给出了HF方法结合不同基组对OC-BH3做GCDA分析的结果,由此我们考察基组对GCDA结果的影响。我们选取了最常用的def2系列基组[20] ,Pople系列基组[18] [35] 以及Dunning相关一致性基组cc-pVnZ系列[19] ,考虑了2-zeta和3-zeta的情况,以及带与不带弥散函数的情况。def2-SVP和def2-TZVP的结果对于d、b、r的差异都相当小。6-31G**与6-311G**之间,以及cc-pVDZ与cc-pVTZ之间,在d、b项上有一定定量上的差异,r项上的差异相对更大一些。虽然从原理上讲,无法说哪种基组下得到的GCDA结果是最合理的,但由于def2-SVP和def2-TZVP的结果相当一致,而且是构建得比较系统、完善的基组,我们可以以其结果作为参照值。对比发现cc-pVTZ的结果也与def2基组的结果很接近,因此可认为是可靠的,而Pople系列基组与之偏差则较大,因此对于GCDA分析的目的至少不是首选。HF、DFT计算对于基组质量要求不很高[36] ,基组达到def2-TZVP、cc-pVTZ的程度时就可以认为已十分理想。由于def2-SVP的结果与def2-TZVP和cc-pVTZ的结果差异可忽略,因此对于大体系推荐用相对廉价的def2-SVP,其计算量与6-31G**相当。而对于小体系若想追求更准确的结果,可以采用def2-TZVP (计算量低于cc-pVTZ)。
从表6中可见,如果给cc-pVnZ系列基组加上弥散函数,即aug-cc-pVnZ系列,GCDA结果完全错误,所得d明显小于b值,导致d-b为负,错误地表明OC-BH3体系的电子是从BH3流向CO,另外弥散函数也相当大地影响了r值。此时不仅总的d、b值不合理,我们还发现很多轨道对d、b的贡献成了明显无意义的较大负值。对6-311G**加上弥散函数成为6-311++G**后,d、b值偏离较为合理的def2-TZVP的结果更远,因此合理性也进一步降低,而且r值更被明显夸大。但相对来说,给6-311G**加弥散函数带来的影响不及cc-pVnZ切换为aug-cc-pVnZ的影响大,一种可能原因是6-311++G**只是增加了s和p壳层的弥散函数,而aug-cc-pVnZ则是对各个角动量都增加了弥散函数,因此对结果的影响明显更大。总之,做GCDA计算绝不应使用带弥散函数的基组,不仅额外浪费了计算量,还可能使分析结果失去化学意义,甚至被结果所误导。之所以弥散函数使GCDA丧失合理性,我们发现很大一部分原因是因为弥散函数导致片段的最低非占据分子轨道(LUMO)延展范围比不加弥散函数时明显更广,这会导致与另一个片段的s原子轨道构成的成键片段轨道发生过度混合。
5.2. 顺式二氯二胺合铂
顺式二氯二胺合铂(顺铂)是典型的配位化合物。对此体系做GCDA分析时我们将PtCl2作为片段1,(NH3)2作为片段2。相较于上一节的OC-BH3,此体系电子结构更为复杂,过渡金属的存在会使得体系电子相关效应更为显著。表7列出了不同方法下对顺铂不同片段间的CDA和GCDA计算结果。由数据可见无论什么级别,给出的基本结论是一致的,即(NH3)2向PtCl2贡献了约0.2~0.3个电子,同时也伴随着少量反馈,但不同级别下得到的d、b、r存在一定定量差别,可以看到d项的大体趋势都是:低HF成份的泛函 > 高HF成份的泛函 > HF ³ MP2 > CCSD,这个趋势与前一节分析OC-BH3的情况基本一致。之
Table 5. GCDA results of OC-BH3 system calculated by various methods. HF exchange composition of DFT functionals are in the parentheses
表5. 不同方法计算的OC-BH3体系的GCDA结果。括号内为DFT泛函的HF交换成份
a范围分离泛函,近程HF成份为22.2%,远程为100%。
Table 6. GCDA results of OC-BH3 system obtained by HF method in combination with different basis-sets
表6. HF方法结合不同基组得到的OC-BH3体系的GCDA结果
表7. 不同方法计算的顺式二氯二胺合铂的GCDA/CDA结果
所以有这样的趋势,可能的原因在于HF成份越低的DFT泛函自相互作用误差越大[36] ,因此复合物电荷分布越倾向于离域,造成占据和非占据片段轨道在复合物轨道中的混合程度越高,从而d项比HF大得越多。HF方法由于交换势是精确的而没有自相互作用误差问题。值得一提的是M06-HF是个例外,尽管它的HF成份高达100%,并且在OC-BH3的例子中和HF的结果十分接近,在此例中却与HF的结果相差显著,由于它的特殊性我们不建议用M06-HF做GCDA计算。在HF基础上考虑了电子相关的MP2方法的d项略小于HF的结果,而且电子相关效应考虑得更充分的CCSD的d项比MP2更小。虽然GCDA中的d、b、r项并不是可观测量,因此无法严格地获得,但由于CCSD是较高级别的后HF方法,有理由将CCSD的结果作为参照值。虽然DFT泛函和MP2、CCSD一样都考虑了动态相关,但是其GCDA结果却与CCSD相差显著,一定程度上表明DFT轨道可能并不很适合用于GCDA的目的。原理上讲,KS-DFT方法引入轨道的初衷是为了获得较准确的动能[37] ,而不是获得合理的波函数,因此轨道的物理意义也并不严格,基于DFT轨道进行的GCDA分析也因而不一定理想,甚至可能不如使用HF轨道,尽管DFT泛函对于计算分子性质,包括电子密度分布等属性几乎总是优于HF。
从表7可见,对于此例CDA和GCDA的结果在CCSD级别下结果大致相同,但是在MP2下差异略为明显。GCDA下MP2的d、d-b值比在CDA下更接近于HF的结果。我们通过ADCH方法计算了(NH3)2配体的片段电荷,HF、MP2和CCSD下分别为0.586、0.588、0.573。虽然如前所述原理上d-b值并不能准确反应电荷转移量,但片段电荷在一定程度上还是表明MP2的d-b项应当与HF的接近,GCDA比CDA的结果更满足这一点。而且由于GCDA对复合物和片段都使用MP2自然轨道,计算级别能够保持统一,因此在后HF级别下建议使用本文提出的GCDA形式。
6. 结论
CDA是重要的深入研究电荷转移问题的方法。本文我们对CDA方法进行了介绍,并对它进行了广义化,提出了GCDA方法。相对于CDA,GCDA方法将片段轨道占据数纳入了考虑,在HF、DFT级别下等价于CDA,而在后HF级别下比CDA形式更为合理,因为此时复合物轨道和片段轨道能够在相同后HF级别下产生,而CDA则要求在HF级别下产生片段轨道。另外GCDA也明确考虑了用于开壳层体系时的情况。我们在文中还对CDA/GCDA计算时应注意的诸多要点做了阐述。
为了考察GCDA方法的特点与合理性,我们以OC-BH3和顺铂体系为实例进行了探究。我们发现GCDA和CDA的结果无论是d、b、r总值上还是复合物轨道的独立贡献上都较为接近,但在个别情况下也会有一定定量差异。若电子静态相关很强,导致片段轨道占据数偏离整数较大,则差异可能会更大,此时应采用定义更合理的GCDA的结果。理论方法对GCDA的结果有明显影响,d项存在大体趋势:低HF成份的泛函 > 高HF成份的泛函 > HF ³ MP2 > CCSD。DFT的GCDA结果无论是从总值上还是复合物轨道独立贡献上都与HF定性上一致。虽然在计算分子性质上DFT明显优于HF,但若以较高精度的CCSD的结果为参照,DFT用于GCDA分析未必优于HF。MP2和CCSD这两种后HF方法不仅在GCDA总结果上相近,其复合物自然轨道产生的独立贡献的特征也相似。虽然我们发现后HF级别下复合物自然轨道的独立贡献与HF级别下的复合物分子轨道的独立贡献明显缺乏可比性,但通过图形考察复合物和片段自然轨道的特征,能够确认后HF级别下GCDA的结果也是合理的。另外,我们也考察了基组对GCDA结果的影响,发现def2-SVP已足矣得到合理的结果。弥散函数绝不应当在GCDA分析时使用,否则将严重破坏结果的合理性。
文章引用
肖 萌,卢 天. 广义化的电荷分解分析(GCDA)方法
Generalized Charge Decomposition Analysis (GCDA) Method[J]. 物理化学进展, 2015, 04(04):
111-124.
http://dx.doi.org/10.12677/JAPC.2015.44013
参考文献 (References)
- 1. 周光耀. 氢键的量子化学研究(一)[J]. 物理化学进展, 2015, 4(2): 84-101.
- 2. 卢天, 陈飞武. 原子电荷计算方法的对比[J]. 物理化学学报, 2012, 28(1): 1-18.
- 3. Reed, A.E., Weinstock, R.B. and Weinhold, F. (1985) Natural Population Analysis. The Journal of Chemical Physics, 83, 735-746. http://dx.doi.org/10.1063/1.449486
- 4. 卢天, 陈飞武. 分子轨道成分的计算[J]. 化学学报, 2011, 69(20): 2393-2406.
- 5. Dapprich, S. and Frenking, G. (1995) Investigation of Donor-Acceptor Interactions: A Charge Decomposition Analysis Using Fragment Molecular Orbitals. The Journal of Physical Chemistry, 99, 9352-9362. http://dx.doi.org/10.1021/j100023a009
- 6. Gorelsky, S.I., Ghosh, S. and Solomon, E.I. (2005) Mechanism of N2O Reduction by the μ4-S Tetranuclear CuZ Cluster of Nitrous Oxide Reductase. Journal of the American Chemical Society, 128, 278-290. http://dx.doi.org/10.1021/ja055856o
- 7. Zhao, M., Wang, L., Li, P., Zhang, X., Yang, Y. and Zheng, W. (2015) Paddlewheel 1,2,4-Diazaphospholide Dibismuthanes with Very Short Bismuth-Bismuth Single Bonds. Chemical Communications, 51, 16184-16187. http://dx.doi.org/10.1039/C5CC07064C
- 8. Lan, J.-H., Wang, C.-Z., Wu, Q.-Y., Wang, S.-A., Feng, Y.-X., Zhao, Y.-L., Chai, Z.-F. and Shi, W.-Q. (2015) A Quasi-relativistic Density Functional Theory Study of the Actinyl(VI, V) (An = U, Np, Pu) Complexes with a Six-Mem- bered Macrocycle Containing Pyrrole, Pyridine, and Furan Subunits. The Journal of Physical Chemistry A, 119, 9178- 9188. http://dx.doi.org/10.1021/acs.jpca.5b06370
- 9. Wagner, A., Kaifer, E. and Himmel, H.-J. (2013) Bonding in Diborane-Metal Complexes: A Quantum-Chemical and Experimental Study of Complexes Featuring Early and Late Transition Metals. Chemistry: A European Journal, 19, 7395-7409. http://dx.doi.org/10.1002/chem.201300348
- 10. Zhu, Y., Day, C.S., Zhang, L., Hauser, K.J. and Jones, A.C. (2013) A Unique Au-Ag-Au Triangular Motif in a Trimetallic Halonium Dication: Silver Incorporation in a Gold(I) Catalyst. Chemistry: A European Journal, 19, 12264-12271. http://dx.doi.org/10.1002/chem.201302152
- 11. Roy, D.K., Mondal, B., Anju, R.S. and Ghosh, S. (2015) Chemi-stry of Diruthenium and Dirhodium Analogues of Pentaborane(9): Synthesis and Characterization of Metal N,S-Heterocyclic Carbene and B-Agostic Complexes. Chemistry: A European Journal, 21, 3640-3648. http://dx.doi.org/10.1002/chem.201405218
- 12. Yao, Y.S., Yong, X., Tse, J.S. and Greschner, M.J. (2014) Dihy-drogen Bonding in Compressed Ammonia Borane and Its Roles in Structural Stability. The Journal of Physical Chemi-stry C, 118, 29591-29598. http://dx.doi.org/10.1021/jp509633h
- 13. Li, Z.-F., Yang, X.-P., Hui-Xue, L. and Guo, Z. (2014) Electronic Structure of Gold Carbonyl Compounds RAuL (R = CF3, BO, Br, Cl, CH3, HCC, Mes3P, SIDipp; L = CO, N2, BO) and Origins of Aurophilic Interactions in the Clusters [RAuL]n (n = 2 - 4): A Theoretical Study. Organometallics, 33, 5101-5110. http://dx.doi.org/10.1021/om4007505
- 14. Das, R. and Chattaraj, P.K. (2014) Gas Storage Potential of ExBox4+ and Its Li-Decorated Derivative. Physical Chemistry Chemical Physics, 16, 21964-21979. http://dx.doi.org/10.1039/C4CP02199A
- 15. Wang, W.-Y., Ma, N.-N., Sun, S.-L. and Qiu, Y.-Q. (2014) Impact of Redox Stimuli on Ferrocene-Buckybowl Complexes: Switchable Optoelectronic and Nonlinear Optical Properties. Organometallics, 33, 3341-3352. http://dx.doi.org/10.1021/om500224g
- 16. Yang, X., Liang, Y.N., Ding, S.D., Li, S.J., Chai, Z.F. and Wang, D.Q. (2014) Influence of a Bridging Group and the Substitution Effect of Bis(1,2,4-triazine) N-Donor Extractants on Their Interactions with a NpV Cation. Inorganic Chemistry, 53, 7848-7860. http://dx.doi.org/10.1021/ic500138w
- 17. Mulliken, R.S. (1955) Electronic Population Analysis on LCAO-MO Molecular Wave Functions. II. Overlap Populations, Bond Orders, and Covalent Bond Energies. The Journal of Chemical Physics, 23, 1841-1846. http://dx.doi.org/10.1063/1.1740589
- 18. Hariharan, P.C. and Pople, J.A. (1973) The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theoretica Chimica Acta, 28, 213-222. http://dx.doi.org/10.1007/BF00533485
- 19. Dunning Jr., T.H. (1989) Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. The Journal of Chemical Physics, 90, 1007-1023. http://dx.doi.org/10.1063/1.456153
- 20. Weigend, F. and Ahlrichs, R. (2005) Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Physical Chemistry Chemical Physics, 7, 3297- 3305. http://dx.doi.org/10.1039/b508541a
- 21. Wadt, W.R. and Hay, P.J. (1985) Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for Main Group Elements Na to Bi. The Journal of Chemical Physics, 82, 284-298. http://dx.doi.org/10.1063/1.448800
- 22. Stephens, P.J., Devlin, F.J., Chabalowski, C.F. and Frisch, M.J. (1994) Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. The Journal of Physical Chemistry, 98, 11623- 11627. http://dx.doi.org/10.1021/j100096a001
- 23. Neese, F. (2012) The ORCA Program System. Wiley Interdisciplinary Reviews: Computational Molecular Science, 2, 73-78. http://dx.doi.org/10.1002/wcms.81
- 24. Lu, T. and Chen, F. (2012) Multiwfn: A Multifunctional Wavefunction Analyzer. Journal of Computational Chemistry, 33, 580-592. http://dx.doi.org/10.1002/jcc.22885
- 25. Website of Multiwfn Program. http://Multiwfn.codeplex.com
- 26. Lu, T. and Chen, F.W. (2012) Atomic Dipole Moment Corrected Hirshfeld Population Method. Journal of Theoretical and Computational Chemistry, 11, 163-183. http://dx.doi.org/10.1142/S0219633612500113
- 27. Becke, A.D. (1988) Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Physical Review A, 38, 3098. http://dx.doi.org/10.1103/PhysRevA.38.3098
- 28. Lee, C., Yang, W. and Parr, R.G. (1988) Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Physical Review B, 37, 785-789. http://dx.doi.org/10.1103/PhysRevB.37.785
- 29. Perdew, J.P., Burke, K. and Ernzerhof, M. (1996) Generalized Gradient Approximation Made Simple. Physical Review Letters, 77, 3865-3868. http://dx.doi.org/10.1103/PhysRevLett.77.3865
- 30. Adamo, C. and Barone, V. (1999) Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. The Journal of Chemical Physics, 110, 6158-6170. http://dx.doi.org/10.1063/1.478522
- 31. Becke, A.D. (1993) A New Mixing of Hartree—Fock and Local Density-Functional Theories. The Journal of Chemical Physics, 98, 1372-1377. http://dx.doi.org/10.1063/1.464304
- 32. Zhao, Y. and Truhlar, D. (2008) The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theoretical Chemistry Accounts, 120, 215- 241. http://dx.doi.org/10.1007/s00214-007-0310-x
- 33. Zhao, Y. and Truhlar, D.G. (2006) Density Functional for Spectroscopy: No Long-Range Self-Interaction Error, Good Performance for Rydberg and Charge-Transfer States, and Better Performance on Average than B3LYP for Ground States. The Journal of Physical Chemistry A, 110, 13126-13130. http://dx.doi.org/10.1021/jp066479k
- 34. Chai, J.-D. and Head-Gordon, M. (2008) Long-Range Corrected Hybrid Density Functionals with Damped Atom- Atom Dispersion Corrections. Physical Chemistry Chemical Physics, 10, 6615-6620. http://dx.doi.org/10.1039/b810189b
- 35. Frisch, M.J., Pople, J.A. and Binkley, J.S. (1984) Self-Consistent Mo-lecular Orbital Methods 25. Supplementary Functions for Gaussian Basis Sets. The Journal of Chemical Physics, 80, 3265-3269. http://dx.doi.org/10.1063/1.447079
- 36. Jensen, F. (2007) Introduction to Computational Chemistry. 2th Edition, John Wiley & Sons, West Sussex.
- 37. Koch, W., Holthausen, M. C. (2001) A Chemist’s Guide to Density Functional Theory. 2th Edition, Wiley-VCH Verlag GmbH, Weinheim. http://dx.doi.org/10.1002/3527600043