Hans Journal of Chemical Engineering and Technology
Vol.07 No.06(2017), Article ID:22899,10
pages
10.12677/HJCET.2017.76043
Conjugated Polycarbazole Network for Gas Storage and Separation
Fei Jiang, Guie Chen, Haifang Mao, Jun Yu, Jing Ye, Zijian Yao, Jianyong Zhang, Wei Deng*
School of Chemical and Environmental Engineering, Shanghai Institute of Technology, Shanghai
*通讯作者。

Received: Nov. 10th, 2017; accepted: Nov. 23rd, 2017; published: Nov. 30th, 2017

ABSTRACT
Two Conjugated Polycarbazole frameworks P-1 (Cz-DTBT-Cz) and P-2 (Cz-TT-Cz) with Z-type DTBT moiety and propeller-type TT bithiophene as the core were synthesized to decrease the emissions of greenhouse gas such as CO2 and so on. The unique structure of DTBT moiety and propeller-type TT bithiophene moiety is a superior candidate to constructed organic porous materials. The results turn out that the performances of obtained polymers are quite different based on the two strategies. Both the thermogravimetric curve shows that the two compounds are very stable. The BET result shows that P1 with a BET of 752 m2∙g−1 is 1.52 folders of P2 (589) m2∙g−1. CO2 isotherms show P1 owns the better gas uptake abilities. The results make it clear that the synthetic path and polymerization methods can make great difference on the performance of materials and the path 1 reported here is much better selection to prepare the P-1 (Cz-DTBT-Cz) with ultra-micropores and brilliant ability for gas adsorption.
Keywords:Conjugated Microporous Polymers, Absorption, Storage, Separation
共轭微孔聚合物在CO2气体存储和分离方面的应用基础研究
姜飞,陈桂娥,毛海舫,俞俊,叶静,姚子健,张建勇,邓维*
上海应用技术大学,化学与环境工程学院,上海

收稿日期:2017年11月10日;录用日期:2017年11月23日;发布日期:2017年11月30日

摘 要
本研究以Z型结构的二己基噻吩并苯并噻二唑(DTBT)单体和螺旋桨结构的连噻吩(TT)为中间核,通过改变单体的空间扭曲度,来增加聚合物的比表面积,设计了两类共轭微孔聚合物P-1 (Cz-DTBT-Cz)和P-2 (Cz-TT-Cz)来用于CO2气体的吸附、储存及分离。热重分析表明两聚合物的稳定性良好;随后测定了纯气体N2的吸附等温线,氮气吸附等温线显示P-1的比表面积(752 m2∙g−1)是P-2 (589 m2∙g−1)的1.52倍,并利用Clausius Clapeyron方程计算出了相关的吸附焓值,测试结果表明,P-1在各个方面的性能都优于P-2,在清洁能源与环境领域具有较大的发展潜力。
关键词 :共轭微孔,吸附,存储,分离

Copyright © 2017 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 引言
随着石油等化石原料的不断使用,使得CO2等温室气体的排放量不断增加,据统计大气中的CO2浓度已经增长到工业革命以前的1.4倍,对人们的生活环境造成了严重的影响 [1] 。因此寻求高效的CO2处理方法,实现CO2的有效捕集,是十分必要的;同时随着以H2、CH4 [2] [3] 为代表的清洁能源 [4] [5] 不断发展,如何实现燃料气的有效存储与分离是人们关注的另一个热点。而共轭微孔聚合物 [6] [7] 凭借其稳定的化学性质、较大的比表面积、均匀窄小的孔径分布以及较低的骨架密度等特点,使其在众多的气体吸收及分离 [8] 等方面有着较好的发展前景。但是由于共轭微孔聚合物的吸收存储性能受到单体空间结构、孔道形貌以及有效的化学基团等多方面的影响,而其中单体的有效结构又是决定微孔聚合物最终吸收性能的决定性因素,因此寻求合适的合成材料,以获得拥有较为良好空间构型的单体,实现CO2的高效的捕集以及H2、CH4等燃料气的存储分离 [9] [10] ,是研究共轭微孔聚合物的有效途径。
咔唑及其衍生物是一类具有刚性稠环结构的含氮芳杂环化合物,该类化合物具有较高的光热稳定性 [11] [12] ,用其搭建的干态微孔道具有永久性,而且结构中的氮杂环可以对CO2等酸性气体起到一定的吸收作用 [13] ,分子中较大的共轭体系和较强的内电荷转移使得咔唑类化合物可以在一定情况下引入某种特定的官能团,而具有一定的可设计性,因此咔唑是合成共轭微孔聚合物的理想材料。而噻吩富电子结构,可以与负电子的CO2之间形成较强的作用 [14] [15] 。此外,由于该类化合物具有π-共轭体系使其通过简单的掺杂或者化学修饰的方法就可以得到不同空间构型的聚合物,也是合成共轭微孔聚合物的理想材料,但纯噻吩类的聚合物刚性较弱,在干燥过程中会使体系发生坍塌,孔道减少。通过将带有稠环结构的咔唑基团引入材料骨架中,可以增强聚合物的刚性,获得较为理想的孔道结构。同时以噻吩为中间核,设计不同空间构型的连噻吩聚合物,可以在一定程度上增大聚合物的空间扭曲度,进而增大微孔材料的比表面积。本实验以咔唑为基底,以共轭性能较好的DTBT和TT为中间核,合成了两种不同空间构型的微孔材料,通过增大单体的空间扭曲度,以期来增加聚合物的比表面积,从而来获得良好的气体吸附能力。
2. 实验材料与设备
2.1. 实验材料
① 噻吩,苯并噻二唑,咔唑(99%),硝基苯,铜粉,N-溴代丁二酰亚胺(NBS) (99%)百灵威科技有限公司;② 乙腈(AR),氯仿(AR),DMF(AR),甲醇(AR),二氯甲烷(AR),无水硫酸钠(AR),无水碳酸钾(AR),氨水(AR),西陇化工股份有限公司;③ 四氢呋喃(THF)(HPLC),无水三氯化铁(97%),N,N-二甲基乙酰胺(DMAc, 99.8%)天津赛孚瑞科技有限公司;④ 硅胶,200-300目,青岛海洋化工试剂公司;⑤ 石油醚,工业级,直接使用;⑥ 氯仿及其干燥:氩气保护下,避光,在氯仿中加入适量的氢化钙,40℃搅拌过夜。常压下蒸馏,收集备用;⑦ THF及其纯化:将色谱纯的THF提前用氢氧化钾预干燥,然后将THF转移至加热回流装置中,氮气保护下,加入金属钠,回流4小时左右后,再加入显色剂二苯甲酮,继续回流,待溶液变为蓝色或深紫色即可蒸出,收集使用。
试剂:所用化学试剂多数为J&K、Aldrich、Acros以及阿拉丁试剂公司,对有特殊要求的反应,所用试剂及溶剂均需经过常规纯化处理过程。
2.2. 实验设备
① 核磁共振仪:Bruker AV 600M核磁共振仪测试单体的1H and 13C NMR谱,tetramethylsilane (TMS)作为内标;② 红外光谱仪:Thermo Nicolet 6700 FT-IR Spectrometer测试聚合物的FT-IR光谱,采用attenuated total reflection (ATR)模式;③ 扫描电子显微镜:Hitachi S-4800冷场发射扫描电子显微镜,测试加速电压3.0 kV,样品测试前喷金处理;④ 热重分析仪:聚合物的稳定性测试通过TQ-600热重分析仪测试,氮气气氛,量程600℃,升温速率5℃/min;⑤ 固体核磁共振仪:聚合物固体核磁13C CP-MAS由固态交叉极化魔角旋转核磁共振仪Bruker AVANCE-III进行测试;⑥ 气体吸附仪:美国康塔仪器公司Autosorb-iQ-MP-VP体积吸附分析仪。
3. 实验方法
3.1. 材料的表征与测试条件
测试单体的1H and 13C NMR光谱由Bruker AV 600 MHz核磁共振仪得出;聚合物固体核磁13C CP-MAS由固态交叉极化魔角旋转核磁共振仪Bruker AVANCE-III 400 MHz进行测试。测试聚合物的FT-IR光谱,由Thermo Nicolet 6700 FT-IR Spectrometer得出;聚合物的稳定性测试通过Q-5000IR热重分析仪测试,氮气气氛,量程20℃~800℃,升温速率10℃/min;Hitachi S-4800冷场发射扫描电子显微镜测试聚合的外观形貌,测试加速电压5.0 kV,样品测试前喷金处理。气体吸附测试:Quantachrome Autosorb-iQ-MP-VP体积吸附分析仪,测试前,样品120℃脱气处理600分钟,聚合物孔道性质通过77 K下0~1.0 bar氮气吸附-脱附等温线研究,其孔径分布由吸附等温线通过淬灭固体密度泛函理论计算得来。CO2吸附等温线在273 K,298 K和0~1.0 bar条件下完成。
3.2. 材料PCz-TT-Cz和PCz-DTBT-Cz的合成
以下所有的反应均在氩气氛围下进行,四氢呋喃,氯仿在使用前,通过蒸馏进行提纯。咔唑因含有一定的杂质在使用前我们利用石油醚为洗脱剂进行了硅胶柱层析进行分离提纯,其他原料以及试剂如无特殊说明均可直接使用。聚合物有机微孔材料PCz-TT-Cz和PCz-DTBT-Cz的具体合成路线如下图1所示,其中连噻吩(2)、二溴连噻吩(3)、DTBT(6)和二溴DTBT(7)均参照文献制备 [3] ,具体实验步骤不再详述。
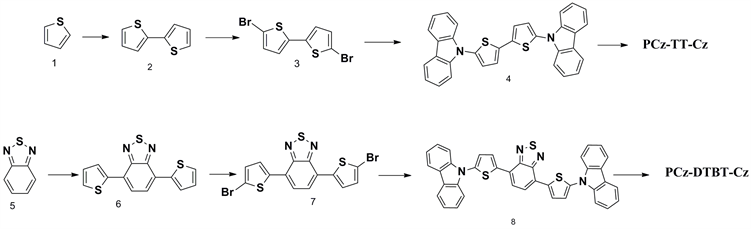
Figure 1. Synthetic pathway towards the materials of PCz-TT-Cz and PCz-DTBT-Cz
图1. 材料PCz-TT-Cz和PCz-DTBT-Cz的合成路线
3.3. 单体Cz-TT-Cz(4)的合成
反应开始前首先将Cu粉进行活化。称取Cu粉(200 mg)放入200mL锥形瓶中,加入5 mL浓HCl,100 mL丙酮,加入磁子搅拌半小时,搅拌结束,将其进行抽滤处理。再次将Cu粉加入到丙酮溶液中,加入三到五粒碘粒继续活化,搅拌半小时后活化完成的红棕色Cu粉抽滤出来作为催化剂进行下一步反应。
氩气保护下,将化合物3 (600 mg, 1.85 mmol)加入到100 mL装有磁子的干燥梨形反应瓶中,依次加入咔唑(928 mg,5.55 mmol),活化的铜粉200 mg,碳酸钾(1.023 g,7.4 mmol)以及20 mL硝基苯。油浴加热到180℃,反应24小时。因硝基苯沸点较高水泵旋蒸去除不掉,因此反应结束后将其冷却至室温,架上蒸馏装置用油泵将硝基苯溶液蒸馏出来,蒸馏结束后室温加入30 mL氨水搅拌3小时以除去催化剂。反应结束,加水加入二氯进行有机相萃取,有机相用无水Na2SO4干燥,旋转蒸发仪旋干溶剂,硅胶粉炒样上样,硅胶柱进行分离提纯。洗脱剂石油醚:二氯甲烷为4:1,收集黄色色带,得到最终黄色粉末的单体产物化合物4 (220 mg, 36.7%)。
3.4. 单体Cz-DTBT-Cz(8)的合成
与单体Cz-TT-Cz合成步骤相似,反应开始前按照之前Cu粉活化方法将其进行活化作为催化剂进行下一步反应。氩气保护下,将化合物7 (570 mg, 1.24 mmol)加入到100 mL装有磁子的干燥梨形反应瓶中,依次加入咔唑(620 mg, 3.72 mmol),活化的铜粉200 mg,碳酸钾(687 mg, 4.96 mmol)以及20 mL硝基苯。使用油泵换气三次。换气结束将反应油浴加热到180℃,反应24小时。因硝基苯沸点较高水泵旋蒸去除不掉,因此反应结束后将其冷却至室温,架上蒸馏装置用油泵将硝基苯溶液蒸馏出来,蒸馏结束后室温加入30 mL氨水搅拌3小时以除去催化剂。反应结束,加水加入二氯进行有机相萃取,有机相用无水Na2SO4干燥,旋转蒸发仪旋干溶剂,硅胶粉炒样上样,硅胶柱进行分离提纯。洗脱剂石油醚:二氯甲烷为4:1,收集紫红色色带,得到最终紫色粉末的单体产物化合物8 (240 mg, 42.0%)。
3.5. 聚合物PCz-TT-Cz的合成
在含有磁子高温干燥过的50 mL高压管中将单体Cz-TT-Cz (200 mg, 0.40 mmol)溶解在30 mL经CaH2干燥后氯仿中,加入三氯化铁(457.2 mg, 2.82 mmol),氮气保护。180℃高温下避光搅拌24小时。反应结束后,将100 mL甲醇加入反应体系终止反应,室温搅拌30分钟,过滤,甲醇,浓盐酸溶液,去离子水依次冲洗三遍,将所得粗产品置于索氏提取器中,甲醇提取24小时,氯仿提取24小时,在用四氢呋喃提取24小时。所得聚合物80℃真空干燥24小时,最终得到黑色粉末的聚合物PCz-TT-Cz。
3.6. 聚合物PCz-DTBT-Cz的合成
在含有磁子高温干燥过的50 mL高压管中将单体Cz-DTBT-Cz (230 mg, 0.37 mmol)溶解在30 mL经CaH2干燥后氯仿中,加入三氯化铁(414 mg, 2.55 mmol),氮气保护。180℃高温下避光搅拌24小时。反应结束后,将100 mL甲醇加入反应体系终止反应,室温搅拌30分钟,过滤,甲醇,浓盐酸溶液,去离子水依次冲洗三遍,将所得粗产品置于索氏提取器中,甲醇提取24小时,氯仿提取24小时,在用四氢呋喃提取24小时。所得聚合物80℃真空干燥24小时,最终得到黑色粉末的聚合物PCz-DTBT-Cz。
4. 实验表征结果与分析
4.1. 样品表征
4.1.1. 固体核磁谱图
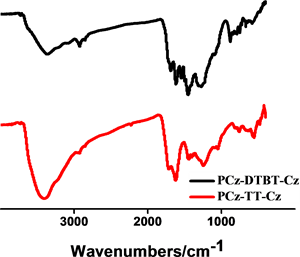
通过FT-IR进一步表征所得的聚合物结构,以材料PCz-DTBT-Cz为例分析图谱(图2),图中722.35 cm−1,878.20 cm−1,1091.20 cm−1,1685.64 cm−1,为噻吩环上的本征吸收峰,749.56 cm−1,1273.77 cm−1,1452.35 cm−1,1617.14 cm−1,为咔唑单元的特征吸收峰。N-S-N,C-N,N-S键的伸缩振动峰在1200到1500之间也均有表现,证明了DTBT基团的存在。两种聚合物材料都表现出噻吩及咔唑的特征吸收峰。有机微孔聚合物材料P-2和P-1的固体碳谱见图2(b),两个聚合物因为都含有咔唑基团,所以均在141 ppm两聚合物均表现出相似的尖峰,具有相似的化学位移。这一结果也与其他文献中 [16] 所报道过的以咔唑为基础的聚合物材料有相似的化学位移。
4.1.2. 扫描电子显微镜图
通过对比可以看到(图3),在相同的放大倍数下这两种聚合物具有完全不同的堆积形貌,P-1为丝状堆积,P-2为类网状堆积,两种聚合物均为无定形堆积形貌。通过扫描电子显微镜图片可以发现以咔唑为基础,中间核体分别引入了共轭性能较好的P-1和P-2为中间核体,可以完全改变聚合物的堆积形貌。
4.1.3. 热重分析
从热重分析图谱(图4)可以看出,P-1和P-2均表现出优良的热稳定性,质量损为5 wt%时,P-2的热分解温度为319℃,P-1的热分解温度为352℃。上述数据表明两种聚合物均有较好的热稳定性。
 (a)
(a)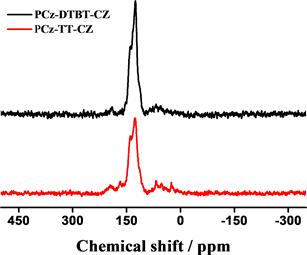 (b)
(b)
Figure 2. (a) FI-IR spectra of PCz-TT-Cz and PCz-DTBT-Cz polymers; (b) Solid state magic angle spinning 13C CP/MAS NMR spectra spectra of PCz-DTBT-Cz and PCz-TT-Cz
图2. (a) 聚合物PCz-DTBT-Cz和PCz-TT-Cz的红外光谱图;(b) PCz-DTBT-Cz和PCz-TT-Cz的固态交叉极化魔角旋转核磁共振谱
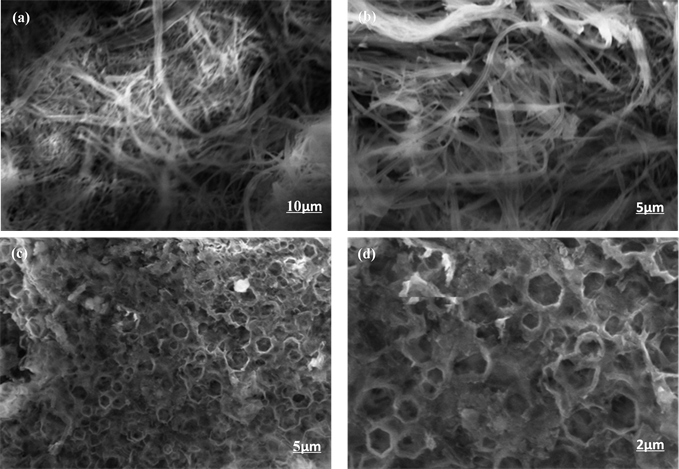
Figure 3. Field-emission scanning electron microscopy images of (a) (b) PCz-DTBT-Cz and (c) (d) PCz-TT-Cz
图3. (a)、(b)和(c)、(d)分别为PCz-DTBT-Cz和PCz-TT-Cz聚合物的扫描电子显微镜图片
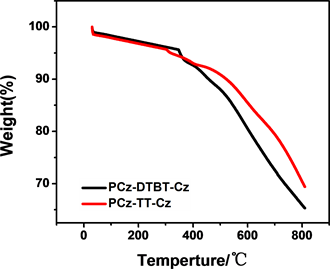
Figure 4. Thermal gravimetric analysis (TGA) curves of polymers PCz-DTBT-Cz and PCz-TT-Cz
图4. 聚合物PCz-DTBT-Cz和PCz-TT-Cz的热失重分析图
4.2. 聚合物气体吸收性能
4.2.1. 氮气吸附等温线
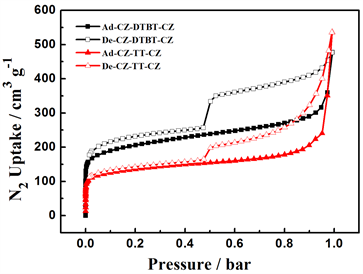
图5中(a)为DTBT和TT为中间核的咔唑类微孔聚合物,在77 K条件下的N2吸附-脱附等温曲线,以氮气为探针分子探测两种聚合物的孔特性。从各吸附等温线的纵坐标N2吸附量可以看出,P-1和 P-2两种聚合物在相对较小的压力小于0.02的低压区,均与N2有较强内吸附作用,氮气吸附量可观,属于
 (a)
(a) 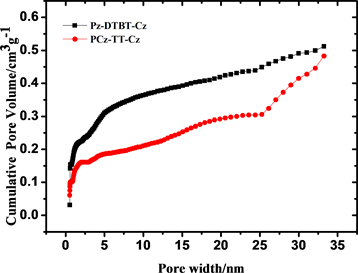 (b)
(b)
Figure 5. (a) Nitrogen adsorption-desorption isotherms of PCz-DTBT-Cz and PCz-TT-Cz Polymers; (b) Different pore volumes were calculated by application of Brunauer-Emmett-Teller (BET)
图5. (a) 77 K条件下聚合物PCz-DTBT-Cz和PCz-TT-Cz的N2吸附-脱附等温线;(b) 淬火固体密度泛函理论计算的PCz-DTBT-Cz和PCz-TT-Cz两种材料的孔容分布
典型的Ⅰ型吸附,说明两种聚合物均具有较大的微孔分布,说明在相同的咔唑基础下,P-1的单体比P-2具有个更大空间扭曲度,进而增大了所得聚合物的比表面积,且前者所形成的孔道结构较稳定、牢固,可以在一定程度上增加材料的骨架刚性保证材料在干态状态下尽可能的保留微孔道结构。而继续增大压力直至0.5 bar左右,在此范围内氮气的吸附量随压力的增加而缓慢增加,表明两种聚合物中含有一定量的介孔分布。在相对较高的压力下,两聚合物对N2的吸附量急剧提升,这是因为在较高压力下,材料中较为疏松的纳米级微孔部分崩塌,形成了小型或边界不清的空隙所造成的 [17] 。采用Brunauer-Emmett- Teller (BET)计算,P-1和P-2的比表面积分别为752 m3/g和589 m3/g,,P-1的比表面积约是P-2的1.52倍,其主要原因可能是前者的中间核在更大程度上改变了单体的空间扭曲度,使得所形成材料的比表面积增大。而且以连噻吩DTBT为中间核所得聚合物的比表面积,高于同类型聚合物的比表面,如以单体噻吩为核,比表面积为577 m3/g [18] ,以螺旋结构的芴为核所得比表面积为450 m3/g [19] ,以芘为核所得比表面积为505 m3/g [20] [21] 。通过对比发现,在氮气脱附实验中,两种聚合物在压力为0.5 bar左右,脱附速率下降速度变快,这一现象主要起源于材料体系内的介孔分布,属于H1滞后环,两种材料体系中可能均存在少量的柱形孔 [22] 。图5中(b)为两聚合物经由淬火固体密度泛函理论计算得出孔容分布图,由图中可以看出,P-1的孔容大于P-2,这可能是中间核DTBT的引入在更大程度上实现了聚合物的空间切割所造成的。
由淬火固体密度泛函理论计算还可得出两种聚噻吩微孔材料的孔径分布曲线,见图6,从图中可以看出聚合物P-1的主孔区分布在0.54 nm和1.00 nm,大于2 nm仅有少量的介孔分布,没有大孔存在;P-2的主孔分布区在0.52 nm和1.00 nm,大于2 nm有一定量的介孔存在,与吸附等温线相吻合。从上述结果可以看出,两种聚合物的孔分布具有一定的均一性,P-1的孔分布范围比P-2更小,孔的分布更加均一,这可能是前者的刚性更强导致的,在一定程度上说明:相同聚合条件下,聚合物骨架刚性的增强有利于孔的均一分布。
4.2.2. CO2吸附等温线
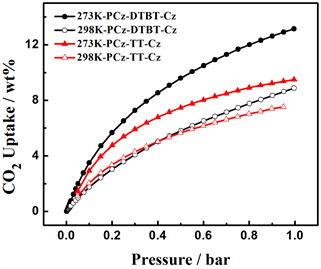
上述N2吸附参数表明,四种噻吩具有不同的孔结构参数,为了进一步研究中间核刚性强弱,空间构型等对聚合物的影响,分别测定了两种聚合物在273 K和298 K条件下的CO2吸附等温线,从图7(a)中
 (a)
(a) (b)
(b)
Figure 6. Different pore size distributions were calculated by application of Brunauer-Emmett-Teller (BET)
图6. 淬火固体密度泛函理论计算的PCz-DTBT-Cz (a)和PCz-TT-Cz (b)两种材料的孔径分布
 (a)
(a) 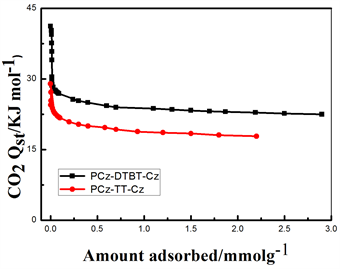 (b)
(b)
Figure 7. (a) CO2 adsorption isotherms of PCz-DTBT-Cz and PCz-TT-Cz Polymers at 273 K and 298 K. (b) CO2 variation of gas isosteric enthalpies with the adsorbed amount
图7. (a) 在273 K和298 K条件下聚合物材料PCz-DTBT-Cz和PCz-TT-Cz的CO2吸附等温线;(b) CO2吸附焓的变化
可以看出在273 K,1bar下P-1的CO2吸附量为13.1 wt%。298 K,1 bar下CO2的吸附量为8.9 wt%。在273 K,1 bar下P-2的CO2吸附量为9.5 wt%。298 K,1 bar下CO2的吸附量为7.7 wt%。从上述结果可以看出,在273 K和298 K,1 bar的条件下两种聚合物均有一定的CO2吸附量,但在273 K、1 bar下,P-1的CO2吸附量是P-2的1.38倍,主要的原因可能是两种聚合物的堆积形貌在一定程度上影响了CO2的吸附量,P-1丝状的堆积结构更有利于CO2分子的附着,此外由于中间核DTBT使得聚合物的空间扭曲度变大,增加了材料的比表面积,两者的共同作用使得CO2吸附能力增强。图7(b)是两种聚合物吸附焓随吸附量变化曲线。从图中可以看出,P-1和P-2在零表面负载时的表面吸附焓分别为29.53 KJ/mol和25.40 KJ/mol,两种聚合物的吸附焓随着表面附载量的不断增加,CO2吸附分子与基底聚合物的作用均呈现下降的趋势,但在整个压力变化范围内,前者的吸附焓都略高于后者,这就可以解释左图中P-1的吸附等温线高于P-2的原因。这可能是在相同的咔唑基础下,相比于中间核TT来说,DTBT的氮硫键拥有较大的电子云密度,可以增强与酸性缺电子结构的CO2之间的相互作用,进而增加CO2的吸附量。从上述CO2的数据结果中可以看出,通过改变中间核的结构,可以提高材料的CO2吸附能力。
5. 结论
本实验合成了两种基于咔唑基团的共轭微孔聚合物P-1 (Cz-DTBT-Cz)和P-2 (Cz-TT-Cz),通过红外光谱和固体核磁证明成功合成了目标产物,利用扫描电镜分析出了两聚合物的具有完全不同的堆积形貌且均为无定形堆积形貌。热重实验分析:P-1和P-2的质量损为5 wt%时,热分解温度均在300℃以上,具有较好的热稳定性;N2吸附试验显示P-1和P-2的比表面积分别为752 m3/g和589 m3/g,前者是后者的1.52倍,其主要原因可能是前者的中间核在更大程度上改变了单体的空间扭曲度,使得所形成材料的比表面积增大。N2吸附试验显示在273 K,1 bar下P-1的CO2吸附量为13.1 wt%,298 K,1 bar下CO2的吸附量为8.9 wt%;在273 K,1 bar下P-2的CO2吸附量为9.5 wt%,298 K,1 bar下CO2的吸附量为7.7 wt%,这一结果与用Clausius Clapeyron方程计算出的相关吸附焓值相一致,且P-1的吸附性能较好,优于报道过的大部分微孔聚合物。综合来看共轭微孔聚合物P-1是更为理想的气体吸收及存储材料。
基金项目
上海市教委项目(ZZyyx15010),上海应用技术大学人才项目(1011XQ171043, DCX2017130, DCX2017136)。
文章引用
姜飞,陈桂娥,毛海舫,俞俊,叶静,姚子健,张建勇,邓维. 共轭微孔聚合物在CO2气体存储和分离方面的应用基础研究
Conjugated Polycarbazole Network for Gas Storage and Separation[J]. 化学工程与技术, 2017, 07(06): 315-324. http://dx.doi.org/10.12677/HJCET.2017.76043
参考文献 (References)
- 1. Trends in Atmospheric Carbon Dioxide, U. S. Department of Commerce, National Oceanic & Atmospheric Administration.
- 2. Rowsell, J.L.C. and Yaghi, O.M. (2005) Strategies for Hydrogen Storage in Metal-Organic Frameworks. Angewandte Chemie International Edition, 44, 4670-4679. https://doi.org/10.1002/anie.200462786
- 3. Murray, L.J., Dinca, M. and Long, J.R. (2009) Hydrogen Storage in Metal-Organic Frameworks. Chemical Society Reviews, 38, 1294-1314. https://doi.org/10.1039/b802256a
- 4. Dawson, R., Cooper, A.I. and Adams, D.J. (2013) Chemical Functionalization Strategies for Carbon Dioxide Capture in Microporous Organic Polymers. Polymer International, 62, 345-352. https://doi.org/10.1002/pi.4407
- 5. Sumida, K., Rogow, D.L., Mason, J.A., McDonald, T.M., Bloch, E.D., Herm, Z.R., Bae, T.H. and Long, J.R. (2012) Carbon Dioxide Capture in Metal-Organic Frameworks. Chemical Reviews, 112, 724-781. https://doi.org/10.1021/cr2003272
- 6. Jiang, F., Sun, J., Yang, R., Qiao, S., An, Z., Huang, J., Mao, H., Chen, G. and Ren, Y. (2016) A Facile Approach to Prepare a Microporous Polycarbazole P-Tetra(4-(N-Carbazolyl)Phenyl)Silane Network with High CO2 Storage and Separation Properties. New Journal of Chemistry, 40, 4969-4973. https://doi.org/10.1039/C5NJ03215F
- 7. Jiang, J.X., Su, F., Trewin, A., Wood, C.D., Campbell, N.L., Niu, H., Dickinson, C., Ganin, A.Y., Rosseinsky, M.J., Khimyak, Y.Z. and Cooper, A.I. (2007) Conjugated Microporous Poly(aryleneethynylene) Networks. Angrewandte Chemie, 46, 8574-8578. https://doi.org/10.1002/anie.200701595
- 8. Preis, E., Widling, C., Brunklaus, G., Schmidt, J., Thomas, A. and Scherf, U. (2013) Microporous Polymer Networks (MPNs) Made in Metal-Free Regimes: Systematic Optimization of a Synthetic Protocol toward N-Arylcarbazole-Based MPNs. ACS Macro Letters, 2, 380-383. https://doi.org/10.1021/mz400126f
- 9. Vilela, F., Zhang, K. and Antonietti, M. (2012) Conjugated Porous Polymers for Energy Applications. Energy & Environmental Science, 5, 7819-7832. https://doi.org/10.1039/c2ee22002d
- 10. Xie, Y., Wang, T.T., Liu, X.H., Zou, K. and Deng, W.Q. (2013) Capture and Conversion of CO2 at Ambient Conditions by a Conjugated Microporous Polymer. Nature Communications, 4, 1960. https://doi.org/10.1038/ncomms2960
- 11. Gu, C., Chen, Y.C., Zhang, Z.B., Xue, S.F., Sun, S.H., Zhang, K., Zhong, C.M., Zhang, H.H., Pan, Y.Y., Lv, Y., Yang, Y.Q., Li, F.H., Zhang, S.B., Huang, F. and Ma, Y.G. (2013) Electrochemical Route to Fabricate Film-Like Conjugated Microporous Polymers and Application for Organic Electronics. Advanced Materials, 25, 3443-3448. https://doi.org/10.1002/adma.201300839
- 12. Liu, X., Xu, Y. and Jiang, D. (2012) Conjugated Microporous Polymers as Molecular Sensing Devices: Microporous Architecture Enables Rapid Response and Enhances Sensitivity in Fluorescence-On and Fluorescence-Off Sensing. Journal of the American Chemical Society, 134, 8738-8741. https://doi.org/10.1021/ja303448r
- 13. Jiang, F., Wang, J., Li, J., Wang, N., Bao, X., Wang, T., Yang, Y., Lan, Z. and Yang, R. (2013) Supramolecular Assemblies with Symmetrical Octahedral Structures—Synthesis, Characterization, and Electrochemical Properties. European Journal of Inorganic Chemistry, 2013, 375-379. https://doi.org/10.1002/ejic.201200923
- 14. Qiao, S., Du, Z., Huang, W. and Yang, R. (2014) Influence of Aggregated Morphology on Carbon Dioxide Uptake of Polythiophene Conjugated Organic Networks. Journal of Solid State Chemistry, 212, 69-72. https://doi.org/10.1016/j.jssc.2013.12.025
- 15. Chen, X., Qiao, S., Du, Z., Zhou, Y. and Yang, R. (2013) Synthesis and Characterization of Functional Thienyl-Phosphine Microporous Polymers for Carbon Dioxi. Macromolecular Rapid Communications, 34, 1181-1185. https://doi.org/10.1002/marc.201300328
- 16. Jiang, F., Wang, N., Du, Z., Wang, J., Lan, Z. and Yang, R. (2012) Thiophene-Coated Functionalized M12L24 Spheres: Synthesis, Characterization, and Electrochemical Properties. Chemistry—An Asian Journal, 7, 2230-2234. https://doi.org/10.1002/asia.201200413
- 17. Kiskan, B. and Weber, J. (2012) ACS Macro Letters, 133, 19416-19421.
- 18. Schmidt, J., Weber, J., Epping, J.D., Antonietti, M. and Thomas, A. (2009) Microporous Conjugated Poly(thienylene arylene) Networks. Advanced Materials, 21,702-705. https://doi.org/10.1002/adma.200802692
- 19. Weber, J. and Thomas, A. (2008) Toward Stable Interfaces in Conjugated Polymers: Microporous Poly(p-phenylene) and Poly(phenyleneethynylene) Based on a Spirobifluorene Building Block. Journal of the American Chemical Society, 130, 6334-6335. https://doi.org/10.1021/ja801691x
- 20. Cheng, G., Hasell, T., Trewin, A., Adams, D.J. and Cooper, A.I. (2012) Soluble Conjugated Microporous Polymers. Angewandte Chemie, 51, 12727-12731. https://doi.org/10.1002/anie.201205521
- 21. Jiang, F., Choy, W.C.H., Li, X.C., Zhang, D. and Chen, J.Q. (2015) Post-treatment-Free Solution-Processed Non-stoichiometric NiOx Nanoparticles for Efficient Hole-Transport Layers of Organic Optoelectronic Devices. Advanced Materials, 27, 2930-2937. https://doi.org/10.1002/adma.201405391
- 22. Budd, P.M., Ghanem, B.S., Makhseed, S., Mckeown, N.B., Msayib, K.J. and Tattershall, C.E. (2004) Polymers of Intrinsic Microporosity (PIMs): Robust, Solution-Processable, Organic Nanoporous Materials. Chemical Communications, 0, 230-231. https://doi.org/10.1039/b311764b