Bioprocess
Vol.2 No.4(2012), Article ID:9241,4 pages DOI:10.4236/BP.2012.24023
Solubilization and Refolding of Inclusion Body Proteins Expressed in Bacterial Host*
State Key Laboratory of Protein and Plant Gene Research, College of Life Sciences, Beijing Normal University, Beijing
Email: #jjing@bnu.edu.cn
Received: Nov. 12th, 2012; revised: Nov. 23rd, 2012; accepted: Dec. 1st, 2012
ABSTRACT:
Inclusion bodies produced in Escherichia coli are composed of densely packed denatured protein molecules in the form of particles. Refolding of inclusion body proteins into bioactive forms is cumbersome, results in poor recovery and accounts for the major cost in production of recombinant proteins from E. coli. With new information available on the structure and function of protein aggregates in bacterial inclusion bodies, it has been possible to develop improved solubilization and refolding procedures for higher recovery of bioactive protein indexed at this paper.
Keywords: Inclusion Body; Solubilization; Protein Refolding; Structure; Biological Activtiy
包涵体溶解与复性的策略与方法*
王 苹,井 健#
北京师范大学生命科学学院,北京市基因工程重点实验室,北京
Email: #jjing@bnu.edu.cn
摘 要:
原核细胞表达外源基因常常会导致包涵体的产生。包涵体的复性是制备有效的功能蛋白质的必需途径。每一种蛋白质的基本结构与性质不同,决定了包涵体中外源蛋白质的复性方法多种多样。本文总结并介绍了目前在包涵体复性方面的几种方法与策略。初步讨论了在复性过程中蛋白质的结构变化。
收稿日期:2012年11月12日;修回日期:2012年11月23日;录用日期:2012年12月1日
关键词:包涵体;蛋白质复性;溶解;蛋白质结构
1. 引言
包涵体是外源基因在宿主细胞中表达时,尤其是在原核细胞中(如大肠杆菌中)高效表达时,形成的一种明显不同于细胞质中的其他成分,它由膜包裹的或无膜裸露的高密度、不溶性的蛋白质颗粒,在光学显微镜下观察可见,为高折射区,多为圆形、卵圆形或不定形等(如图1所示)[1]。包涵体的形成除了与重组蛋白质本身的结构特征有关外,还被认为与宿主菌的培养条件,如培养基成分、温度、pH值、离子强度等因素有关。大肠杆菌中包涵体的基本成分是密集堆积的变性蛋白分子。目前有观点认为,在包涵体内部聚集的变性蛋白质分子有着类天然蛋白的二级结构。
提取包涵体所含有的外源蛋白质分子,首先必须采取措施溶解包涵体中的变性蛋白,进而将其完整的高级空间结构进行恢复,也就是实施复性操作的过程。将包涵体蛋白复性为完全的生物活性态是有难度的。低效率的复性也是许多外源基因在大肠杆菌中的表达产物难以有效制备或者造成损失的主要原因。尽管大肠杆菌表达外源基因时常会导致包涵体的形成并对后期重组蛋白的制备产生困难,但大肠杆菌表达系统仍然是目前重组蛋白生产的一条重要途径。
归其原因,可以看到,虽然包涵体的形成对下游
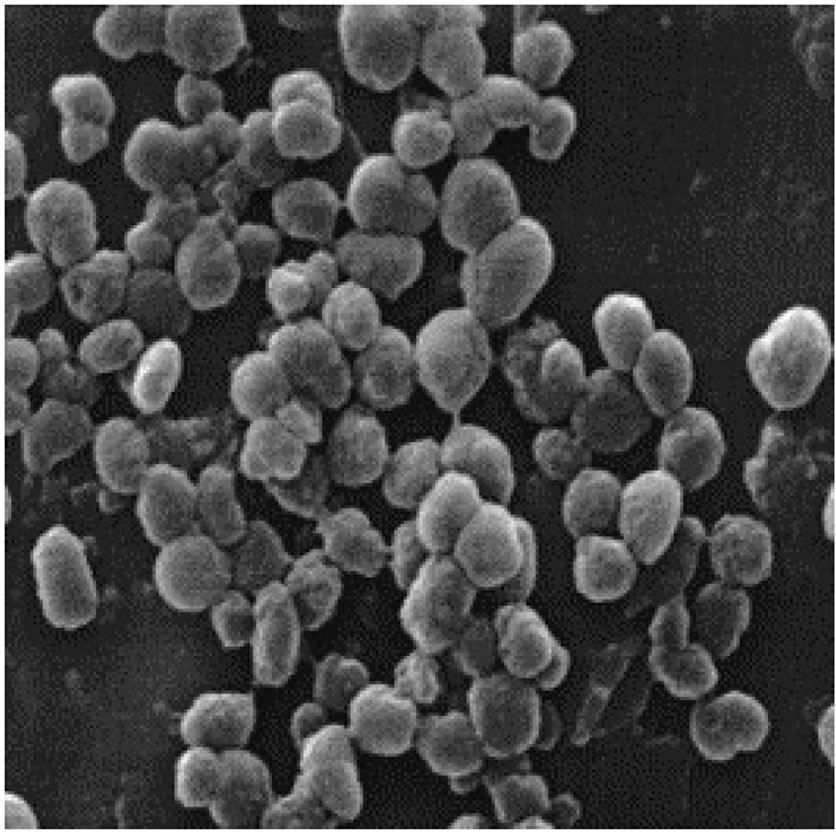
Figure 1. Scanning electron micrograph of inclusion bodies. Diameter of spherical size particle is around 0.5 - 0.8 μm.
图1. 大肠杆菌表达包涵体扫描电镜照片。其中,包涵体球体颗粒的直径在0.5~0.8 uM之间。
纯化与制备造成了困难,但包涵体的形成并非一无是处,在保持蛋白质稳定性以及表达特殊蛋白质方面,却体现出一定的优势。
2. 包涵体表达重组蛋白的优势
为了研究基因的功能,可以把体内的基因转移到其他表达宿主中,研究基因表达性状以确定基因的功能。但是,外源基因的表达,特别是真核生物基因在大肠杆菌中的表达,由于宿主菌缺乏此种基因表达的调控机制,细胞内的化学环境与其天然环境差异,如氧化还原环境、胞内pH、自由水的浓度,以及外源基因的导入,使得表达目的蛋白质的细胞在非正常状态下生长,表达的蛋白质多数以无活性的包涵体的形式存在。在下游生产过程中特别是在医药生产中,以包涵体形式表达的蛋白质具有一些优越性。
1) 异源表达的蛋白质很容易被宿主细胞内的蛋白质酶降解,如果重组蛋白质以包涵体形式表达,由于包埋了酶攻击的位点,可以最大限度地抵抗蛋白质酶的攻击;
2) 包涵体表达的蛋白质没有活性,细胞破碎和以后的纯化步骤不用考虑蛋白质的失活问题;
3) 包涵体形式表达的蛋白质与宿主细胞的其他蛋白质的分离比可溶蛋白质的分离方法简便,造价低,使用简单的离心或者过滤的手段就能使包涵体与宿主细胞的其他蛋白质成分进行有效的分离;
4) 有些表达的外源蛋白质对细胞有毒性或者致死,大量表达时会导致细胞的死亡,最终的细胞数量和产物相当低;而包涵体形式的蛋白质由于丧失了生物活性从而可以高效大量地表达;
5) 对于以包涵体形式表达的产物可以比较容易进行在线观测定性,含有包涵体蛋白质的细胞有较大的折射率,不必像可溶的蛋白质需要细胞破碎后使用酶学或者电泳学的方法进行鉴定。
3. 影响包涵体形成的外部条件及措施
包涵体的形成的原因是复杂的,诸多外部因素都可能影响到包涵体的形成。通过外部条件的调整,有可能降低大肠杆菌包涵体的形成。
1) 降低重组菌的生长温度,降低培养温度是减少包涵体形成的最常用的方法,较低的生长温度降低了无活性聚集体形成的速率和疏水相互作用,从而可减少包涵体的形成。
2) 添加可促进重组蛋白质可溶性表达的生长添加剂,培养E. coli时添加高浓度的多醇类、蔗糖或非代谢糖可以阻止分泌到周质的蛋白质聚集反应,在最适浓度范围内添加这些添加剂不会影响细胞的生长、蛋白质的合成或运输,其它促重组蛋白质可溶性表达的生长添加剂还有乙醇(诱导热休克蛋白的表达)、低分子量的巯基或二硫化合物(影响细胞周质的还原态,从而影响二硫键的形成)和NaCl。
3) 供给丰富的培养基,创造最佳培养条件,如供氧、pH等。
4. 包涵体的洗涤
为了除去包涵体上粘附的杂质,如膜蛋白或核酸,应用洗涤液洗涤包涵体,通常用低浓度的变性剂,过高浓度的尿素或盐酸胍会使包涵体溶解,如2 M尿素在50 mM Tris pH 7.0~8.5左右,1 mM EDTA中洗涤。此外可以用温和去垢剂TritonX-100洗涤去除膜碎片和膜蛋白。
通常的洗涤方法一般是洗不干净的,可以先把包涵体用6 M盐酸胍溶解充分,过滤除去未溶解的物质,注意留样跑电泳,然后用水稀释到4 M,离心把沉淀和上清分别跑电泳,如此类推可以一直稀释到合适的浓度,这就是梯度沉淀的方法,比通常的直接洗脱效果好。
包涵体一般难溶解,所以你要注意未溶解的部分,你可以跑电泳对比,因为有时候难溶解的就是你的目标蛋白,所以每次处理都要把上清和沉淀跑电泳对比,免得把目标蛋白弄丢了。此外刚处理完的包涵体好溶解。冷冻后难溶解,溶解也需要长点时间,也需要大量的溶剂[2]。
5. 包涵体的复性策略
5.1. 透析、稀释和超滤复性法
这三种方法是最传统也是应用最普遍的蛋白质折叠复性方法,复性活性回收率低,而且难于与杂蛋白分离。
透析法很适合实验室应用,好处是不增加体积,通过逐渐降低外透液浓度来控制变性剂去除速度,有人称易形成无活性蛋白质聚体,而且时间长,不适合大规模操作,无法应用到生产规模。另外,如果蛋白质右两个以上的2硫键,无活性蛋白质聚集体,很可能错误形成配对,应用还原剂。
超滤法在膜上聚集变性,在生产中较多的使用,规模较大,易于对透析速度进行控制,缺点是不适合样品量较少的情况,且有些蛋白可能在超滤过程中不可逆的变性,易造成膜污染。
稀释法是直接加入水或缓冲液,放置过夜,缺点是体积增加较大,变性剂稀释速度太快,不易控制,不利于工业放大。目前稀释法主要有一次稀释、分段稀释和连续稀释三种方式[3]。
5.2. 凝胶过滤层析复性
凝胶过滤层析复性又称体积排阻复性(SEC),是一种广泛应用的层析技术。与常用的稀释复性法相比,凝胶过滤层析复性能在高的起始蛋白浓度下对蛋白进行复性,活性回收率较高,同时又能使目标蛋白得到一定程度的纯化。
凝胶过滤复性时,除了蛋白质在胶粒中的传质和扩散外,蛋白质与介质之间并不发生其它任何作用。复性过程始终发生在溶液中。蛋白质在伸展状态中,每个蛋白质分子的伸展状态都会有差别,而不同伸展状态的蛋白质分子在凝胶颗粒内部扩散所受到的限制也不相同,有的会扩散入颗粒内部深一些,有的会浅一些,这使不同伸展状态的蛋白质分子达到一定程度的分离,这样蛋白质分子间相互作用的机会就大大减少,从而起到一定抑制凝集的作用;即使发生了部分凝集,凝集的蛋白质会附着在胶粒上,而不随着溶液向下运动,这样后面的变性剂可以赶上凝集的蛋白质,使其重新溶解并变性;并且在凝胶过滤中脲等变性剂脱除得相对较慢,这对有些蛋白质的复性是有利的。
在普通凝胶过滤中,变性剂和还原剂的脱除虽较稀释复性慢,但变性蛋白质仍会经历变性剂浓度突然变化的过程。脲梯度复性是样品上柱前用复性缓冲液平衡柱子,接着使变性剂浓度递减(如从6 M盐酸胍或8 M尿素下降到复性缓冲液中预先确定的变性剂的浓度)。此法为蛋白质复性提供了一个较温和的环境,实现了线性去除变性剂,对高浓度变性蛋白质的复性效果十分显著。对初始浓度为17 mg/mL的还原变性溶菌酶,在40 min内可以得到高达90%的活性回收率。此外在脲梯度SEC的基础上发展了pH和脲浓度双梯度SEC法用于蛋白复性,效果很好。
在SEC复性的基础上设计的连续基质辅助蛋白复性系统能使变性蛋白定量地转换成复性的天然态蛋白。此复性方法能够将复性过程中产生的蛋白聚集体再次复性,使蛋白最大程度地得到回收。伴侣溶剂塞(Chaperon solvent plug)SEC复性法在一定程度上解决了变性蛋白进入柱顶端之前发生沉淀这一问题。此外,抗体、小分子添加剂如L-精氨酸和环糊精等共价结合到SEC固定相上也可能会提高某些蛋白的复性效率,同时又能使这些物质得以重复利用,降低生产成本[4]。
5.3. 高蛋白浓度下的复性方法
一个成功的复性过程在于能够在高蛋白浓度下仍能得到较高的复性率。一个方法是把变性蛋白缓慢连续或不连续地加入到复性缓冲液中。使得蛋白质在加入过程中或加入阶段之间有足够的时间进行折叠复性,这是由于完全折叠的蛋白通常不会与正在折叠的蛋白一起聚集。第二种方法是用温度跳跃策略。即让蛋白质先在低温下折叠复性以减少蛋白质聚集的形成,当形成聚集体的中间体已经减少时,迅速提高温度以促进蛋白质折叠复性,第三种方法是复性在中等的变性剂浓度下进行,变性剂浓度应高到足以有效防止聚集,同时又必须低到能够引发正确复性。
5.4. 添加促进剂的复性方法
包涵体蛋白质折叠复性促进剂的促进作用可以分为:稳定正确折叠蛋白质的天然结构、改变错误折叠蛋白质的稳定性、增加折叠复性中间体的溶解性、增加非折叠蛋白质的溶解性。通常使用的添加剂有:a) 共溶剂:如PEG6000-20000,通过与中间体特异的形成非聚集的复合物,可以阻止蛋白质分子间的相互碰撞机会,减少蛋白质的聚集。b) 去污剂及表面活性剂:如Trition X-100、CHAPs、磷脂、磺基甜菜碱等对蛋白质复性有促进作用,但它们能与蛋白质结合,很难去除。c) 氧化–还原剂:对于含有二硫键的蛋白,复性过程中应加入氧化还原体系,如GSH/ GSSG、DTT/GSSG、DTE/GSSG等,氧化还原系统通过促进不正确形成的二硫键快速交换反应,提高了正确配对的二硫键的产率。d) 小分子的添加剂:如盐酸胍或尿素、烷基脲、碳酸酰胺类等,都可阻止蛋白聚集,它们的作用可能为:稳定蛋白的活性状态、降低非正确折叠的稳定性、增加折叠中间体的稳定性、增加解折叠状态的稳定性。0.4~0.6 M 的L-Arg:L-Arg能使得不正确折叠的蛋白质结构以及不正确连接的二硫键变得不稳定,使折叠向正确方向进行,可大幅度地提高包涵体蛋白质的折叠效率。e) 添加分子伴侣和折叠酶:分子伴侣是指能够结合和稳定另外一种蛋白质的不稳定构象,并能通过有控制的结合和释放,促进新生多肽链的折叠、多聚体的装配或降解及细胞器蛋白的跨膜运输的一类蛋白质。折叠酶有二硫键异构酶、脯氨酸异构酶等。分子伴侣和折叠酶都能在体外调节蛋白质的正确折叠,提高蛋白质的合成效率。但这类蛋白在折叠复性后要除去,而且十分昂贵,因此采用可回收利用的方法如固定化法为好。f) 人工伴侣:为模仿分子伴侣而发展的一种方法:首先,变性蛋白被复性液中的去污剂捕获,形成蛋白–去污剂复合体,复合体的形成抑制了蛋白的聚集,然后加入环糊精从复合体中去除去污剂,使得蛋白质正确折叠。g) 单克隆抗体:待折叠复性的蛋白质的抗体可有效协助复性,但只限于此蛋白才能获得明显的助折叠作用。h) 其它:多聚离子化合物如肝素可以促进蛋白质的复性,具有稳定天然蛋白质的作用;甘油可以增加黏度,减少分子碰撞的机会,减少错配以提高复性效率;适量的盐浓度可以降低某些带电基团间的斥力,有利于蛋白质的折叠;辅助因子、短链醇、高渗物等能有效的降低聚集体的形成,对蛋白有稳定的作用[5-7]。
5.5. 液相色谱(LC)复性法
液相色谱是一种最有效的纯化蛋白质的方法,已成为基因重组蛋白质纯化的必不可少的手段,现有报道,疏水相互作用色谱(HIC)、离子交换色谱(IEC)、凝胶排阻色谱(SEC)、亲和色谱(AFC)已成功的对变性蛋白进行了复性。与传统的稀释法和透析法相比,液相色谱复性的优点是:a) 在进样后可很快的除去变性剂;b) 由于色谱固定相对变性蛋白质的吸附可明显的减少、甚至完全消除变性蛋白质分子在脱离变性剂环境后的分子聚集,从而避免了沉淀的产生,提高蛋白质复性的质量和活性回收率;c) 在蛋白质复性的同时,可使目标蛋白质与杂蛋白分离达到纯化的目的,使复性和纯化同时进行;d) 便于回收变性剂,降低废水处理成本。
四种色谱法中,SEC的分离效果是LC类最差的,盐酸胍会在IEC柱上保留,与蛋白一起洗脱下来,AFC使用范围窄、所需时间长、价格昂贵,HIC是其中较为理想的。变性蛋白在HIC上的复性机理为:当蛋白质、变性剂和杂蛋白进入HIC系统后,由于变性剂在柱子上的作用力较弱,变性蛋白质的作用力较强,变性剂首先同变性的蛋白质分离,随流动相一起流出色谱柱,又因HIC固定相能提供较常法高出十至数百倍的能量,在变性蛋白质被HIC固定相吸附的同时瞬时除去以水合状态附着在蛋白质表面和与固定相表面接触区域的小分子,而蛋白质的特定的疏水性氨基酸残基与HIC固定相表面作用以形成区域立体结构,接着形成折叠中间体,随着流动相的不断变化,变性蛋白质不断地在固定相表面上进行吸附-解吸附-再吸附,并在此过程中逐渐被复性,形成与天然蛋白质构象相同的蛋白质,并流出色谱柱。HIC固定相是从高盐溶液中吸附变性蛋白质,且与变性剂瞬时分离,不仅大大降低了蛋白质间的聚集作用,还因固定相在分子水平上为变性蛋白提供里很高的能量,使水化的变性蛋白质瞬时失水,并形成局部结构以利于蛋白质从疏水核开始折叠。HIC在蛋白质复性的同时还能与其它杂蛋白进行很好的分离,且HIC柱便宜、快速,故有很好的发展潜力。
5.6. 反胶束复性法
由于蛋白质在反胶束内水相中可以保持其构象和活性,运用相转移技术可以将蛋白质分子包于反胶束内,由于这样可使蛋白质相互分离,减少了蛋白质折叠过程中的聚集作用,通过逐渐降低变性剂的浓度和加入氧化-还原剂,可使变性蛋白质复性,但表面活性剂对蛋白质具有变性作用。
5.7. 双水相复性法
Wetlaufer[8]用硫氰化钠、氯化钠、溴化锂与聚乙二醇构成的双水相系统使得包涵体的溶解与蛋白质的折叠复性在一步双水相技术操作中完成。由于PEG具有稳定蛋白质构象的作用、高浓度盐则具有去稳定的作用,这样正确折叠的蛋白质会不断进入到另一相中,直到蛋白质的折叠与去折叠达到一个平衡。
6. 对于重组蛋白质复性效果的检测方法
根据具体的蛋白性质和需要,可以从生化、免疫、物理性质等方面对蛋白质的复性效率进行检测。
1) 凝胶电泳:一般可以用非变性的聚丙烯酰胺凝胶电泳可以检测变性和天然状态的蛋白质,或用非还原的聚丙烯酰胺电泳检测有二硫键的蛋白复性后二硫键的配对情况。
2) 光谱学方法:可以用紫外差光谱、荧光光谱、圆二色性光谱(CD)等,利用两种状态下的光谱学特征进行复性情况的检测,但一般只用于复性研究中的过程检测。
3) 色谱方法:如IEX、RP-HPLC、CE等,由于两种状态的蛋白色谱行为不同而有所区别。
4) 生物学活性及比活测定:一般用细胞方法或生化方法进行测定,较好的反映了复性蛋白的活性,值得注意的是,不同的测活方法测得的结果不同,而且常常不能完全反映体内活性。
5) 粘度和浊度测定:复性后的蛋白溶解度增加,变性状态时由于疏水残基暴露,一般水溶性很差,大多形成可见的沉淀析出。
6) 免疫学方法:如ELISA、WESTERN等,特别是对结构决定簇的抗体检验,比较真实的反映了蛋白质的折叠状态[9]。
参考文献 (References)
[1] S. M. Singh, A. K. Panda. Solubilization and refolding of bacterial inclusion body proteins. Journal of Bioscience Bioengineering, 2005, 99(4): 303-310.
[2] J. N. Liu, W. Tang, Z. Y. Sun, W. Kung, R. Pannell, P. Sarmientos and V. Gurewich. A site-directed mutagenesis of prourokinase which substantially reduces its intrinsic activity. Biochemistry, 1996, 35: 14070-14076.
[3] J. F. Tait, S. Engelhardt, C. Smith and K. Fujikawa. Prourokinase-annexin V chimeras. Construction, expression, and characterization of recombinant proteins. The Journal of Biological Chemistry, 1995, 270(37): 21594-21599.
[4] A. Constans. Protein purification: Affinity tags II. The Scientist, 2002, 16(4): 37-40.
[5] 王涛, 周先碗, 胡美浩. 人尿激酶原在大肠杆菌中表达的研究[J]. 北京大学学报(自然科学版), 2000, 36(6): 802-807.
[6] H. Lilie, E. Schwarz and R. Rudolph. Advances in refolding of proteins produced in E. coli. Current Opinion in Biotechnology, 1998, 9(5): 497-501.
[7] M. E. Goldberg. Non-detergent sulphobetaines: A new class of molecules that facilitate in vitro protein renaturation. Folding and Design, 1996, 1(1): 21-27.
[8] Y. Xie, D. B. Wetlaufer. Control of aggregation in protein refolding: The temperature-leap tactic. Protein Science, 1996, 5(3): 517-523.
[9] J. G. Thomas, A. Ayling and F. Baneyx. Molecular chaperones, folding catalysts, and the recovery of active recombinant proteins fom E. coli: To fold or refold. Applied Biochemistry and Biotechnology, 1997, 66(3): 197-238.
NOTES
*资助信息:中央高校基本科研业务费专项资金资助项目。
#通讯作者。