Applied Physics
Vol.4 No.08(2014), Article ID:14018,6 pages
DOI:10.12677/APP.2014.48018
Ag Incorporation on ZnO(10 0) Surface: First
Principles Study
0) Surface: First
Principles Study
1Department of Electronics and Information Engineering, Hubei Engineering Institute, Huangshi
2Key Laboratory for Optoelectronics and Communication of Jiangxi Province, Jiangxi Science & Technology Normal University, Nanchang
Email: chenlanli@126.com
Copyright © 2014 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received: Aug. 10th, 2014; revised: Aug. 25th, 2014; accepted: Aug. 27th, 2014
ABSTRACT
Based on the density function theory, we have performed first principles calculations
of energetic stability and conductive properties and electronic structure of Ag
incorporation on ZnO (100) surface, and after
calculations, we have analyzed the relaxation of the structure and formation energy
of Ag incorporation on ZnO (10 0) surface at different
layers. Our results show that compared with pure ZnO (10
0) surface at different
layers. Our results show that compared with pure ZnO (10 0), there is an obvious
effect on the relaxation of the structure for Ag incorporation on ZnO (10
0), there is an obvious
effect on the relaxation of the structure for Ag incorporation on ZnO (10 0) surface. Simultaneously,
we found that the formation energy of the Ag incorporation on the first layer is
the lowest in all cases. Therefore, Ag incorporation on the first layer is the most
stable, which indicates that Ag atom prefers to collect in the surface layer instead
of the bulk. And we found that the system for Ag incorporation on ZnO (10
0) surface. Simultaneously,
we found that the formation energy of the Ag incorporation on the first layer is
the lowest in all cases. Therefore, Ag incorporation on the first layer is the most
stable, which indicates that Ag atom prefers to collect in the surface layer instead
of the bulk. And we found that the system for Ag incorporation on ZnO (10 0) surface demonstrates
as p-type, which is in favor of fabricating p-type material. However, the ionization
of Ag incorporation on ZnO (10
0) surface demonstrates
as p-type, which is in favor of fabricating p-type material. However, the ionization
of Ag incorporation on ZnO (10 0) surface is much
higher, which hinders the electronic ionize.
0) surface is much
higher, which hinders the electronic ionize.
Keywords:Incorporation, First-Principles, Electronic Structure, ZnO Surface

Ag嵌入ZnO(100)表面的第一性原理研究
胡宏铎1,陈兰丽1,2
1湖北工程职业学院信息工程系,黄石
2江西科技师范大学江西省通信与光电子重点实验室,南昌
Email: chenlanli@126.com
收稿日期:2014年8月10日;修回日期:2014年8月25日;录用日期:2014年8月27日

摘 要
基于密度泛函理论,采用第一性原理计算了Ag嵌入ZnO(10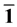 0)面的几何结构和导电性以及电子结构,分析了Ag掺杂ZnO(10
0)面的几何结构和导电性以及电子结构,分析了Ag掺杂ZnO(10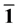 0)面的表面结构弛豫和Ag嵌入ZnO (10
0)面的表面结构弛豫和Ag嵌入ZnO (10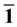 0)不同层的形成能情况。计算结果表明:和纯净ZnO(10
0)不同层的形成能情况。计算结果表明:和纯净ZnO(10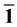 0)相比,Ag嵌入ZnO(10
0)相比,Ag嵌入ZnO(10 0)受结构弛豫影响比较明显。同时发现,Ag嵌入ZnO(10
0)受结构弛豫影响比较明显。同时发现,Ag嵌入ZnO(10 0)第一层的形成能最低,因此,Ag嵌入在第一层的情况最稳定,这表明Ag原子更容易集中在表面层,而不是占据体内位置。通过对纯净和掺杂体系的态密度图分析发现,Ag嵌入ZnO(10
0)第一层的形成能最低,因此,Ag嵌入在第一层的情况最稳定,这表明Ag原子更容易集中在表面层,而不是占据体内位置。通过对纯净和掺杂体系的态密度图分析发现,Ag嵌入ZnO(10 0)表面表现为p型特征,有利于p型的制备。然而,Ag嵌入ZnO(10
0)表面表现为p型特征,有利于p型的制备。然而,Ag嵌入ZnO(10 0)第一层的离化能较高,不利于p型导电。
0)第一层的离化能较高,不利于p型导电。
关键词
嵌入,第一性原理,电子结构,ZnO表面

1. 引言
ZnO光电材料因具有广泛应用领域而受到高度重视。然而,随着研究的进一步深入,发现获得高质量稳定的p型ZnO一直处于瓶颈状态。主要是由于纯净ZnO表现为n型特征,而由于自补偿作用、高离化能,以及低的溶解度,导致p型ZnO薄膜的制备相对困难[1]
。因此,很多研究组采用很多不同的方法去解决这个问题。首先,很多研究组采用单掺的方法得到p型ZnO。例如:Ag[2] 和N[3] 。后来很多的研究小组将问题推向对表面的研究。Tae[4]
等人研究发现Ag原子掺入ZnO表面中,Ag原子占据Zn形成施主能级,而且这种掺杂方式可以改变ZnO薄膜材料的光电性质。Fan[5] [6] 等人发现Ag掺杂ZnO薄膜可以制备压敏电阻、光催化剂和低辐射涂层。Xue[7]
等人报道采用射频反应磁控溅射计算在玻璃衬底上制备Ag掺杂ZnO薄膜材料。研究发现Ag掺杂对ZnO薄膜使得光吸收边向短波长方向移动,但随着Ag浓度的增加又转向长波方向。Duan[8]
等人通过制备Ag掺杂ZnO薄膜发现,ZnO薄膜与Ag相互作用会产生等离子体共振,使得Ag的局域场的强度急剧增强从而使得ZnO薄膜的紫外发光效应增强。正由于Ag掺杂ZnO薄膜可以得到很多奇特的功能,所以一直是人们研究的热点之一[9]
。但是,由于现阶段的工艺条件的限制,纯净ZnO的表面难以制备,而且其稳定性难以保证,从而使得Ag嵌入ZnO(10 0)表面的作用机理研究甚少。
0)表面的作用机理研究甚少。
本文采用第一性原理基于密度泛函理论从理论上对ZnO(10 0)表面的几何结构和电子结构进行了优化计算。计算结果得到了ZnO(10
0)表面的几何结构和电子结构进行了优化计算。计算结果得到了ZnO(10 0)表面的结构弛豫和该表面的导电性能。通过对Ag原子在ZnO(10
0)表面的结构弛豫和该表面的导电性能。通过对Ag原子在ZnO(10 0)表面不同嵌入位置的形成能进行对比分析,得出了Ag嵌入ZnO(10
0)表面不同嵌入位置的形成能进行对比分析,得出了Ag嵌入ZnO(10 0)表面的导电机理。
0)表面的导电机理。
2. 计算方法和计算模型
所有的计算工作都是基于密度泛函理论的第一性原理计算程序即在Vienna Abinit Simulation Package (VASP)[10] [11] 代码采用密度泛函理论。采用的交换关联势为广义梯度近似(GGA)。计算参数设置情况:对表面布里渊区的用Monkhotst-Pack方法取4 × 4 × 1 K点进行积分,平面波的截断能为400 eV。所有的原子全部弛豫直到原子之间的力低于0.01 eV/Å计算停止。所有的计算最下面2层ZnO分子固定,再进行弛豫。通过第一性原理在总能最小的情况选择最优化结构。为了得到可靠的结果,在结构优化基础上进行静态计算。
在本文中,我们首先构建了2 × 2的ZnO(10 0)表面。纯净ZnO(10
0)表面。纯净ZnO(10 0)表面用一块由6层ZnO组成的薄片(slab)来模拟。薄片由包含24个ZnO分子(每层4个)的原胞沿平行于表面的方向无限周期性展开而得,薄片上方有10
Å的真空,以消除平板两端表面之间的相互作用。如图1所示。其中,第一层、第二层、三层、四层、五层以及六层分别标有1、2、3、4、5、6。第一层为表面层;第二层、第三层、第四层为中间层;在这里,第五层和第六层固定,并且模拟为它们处于最优位置的块体环境。而其它四层是完全弛豫。Ag嵌入ZnO(10
0)表面用一块由6层ZnO组成的薄片(slab)来模拟。薄片由包含24个ZnO分子(每层4个)的原胞沿平行于表面的方向无限周期性展开而得,薄片上方有10
Å的真空,以消除平板两端表面之间的相互作用。如图1所示。其中,第一层、第二层、三层、四层、五层以及六层分别标有1、2、3、4、5、6。第一层为表面层;第二层、第三层、第四层为中间层;在这里,第五层和第六层固定,并且模拟为它们处于最优位置的块体环境。而其它四层是完全弛豫。Ag嵌入ZnO(10 0)表面位置如图1所示。Ag掺杂在第一层(如A位置)即为模型1(model
1);Ag掺杂第二层(如B位置)记为模型2(model 2);Ag掺杂第三层(如C位置)记为模型3(model 3);Ag掺杂第四层(如D位置)记为模型4(model
4)。
0)表面位置如图1所示。Ag掺杂在第一层(如A位置)即为模型1(model
1);Ag掺杂第二层(如B位置)记为模型2(model 2);Ag掺杂第三层(如C位置)记为模型3(model 3);Ag掺杂第四层(如D位置)记为模型4(model
4)。
3. 结果与讨论
3.1. 几何结构
弛豫后,表面二聚体出现较大扭曲(如图2所示)。计算结果如表1所示,结合表1和图2发现:和纯净ZnO(10 0)表面相比,Ag嵌入在第一层时的Zn-O健长大约为1.939 Å比纯净中的Zn-O健要长。Zn-O-Zn和O-Zn-O的健角比纯净的要小,呈现出压缩状态。对于model
2,Ag掺杂在第二层时的Zn-O健长大约为1.864 Å,和纯净中的Zn-O健要相差不大。而Zn-O-Zn的健角减小得比较多。对于model 3,Zn-O健长为1.861
Å,O-Zn-O健角为105.006˚,Zn-O-Zn健角为113.003˚。其中健长变短,而O-Zn-O健
0)表面相比,Ag嵌入在第一层时的Zn-O健长大约为1.939 Å比纯净中的Zn-O健要长。Zn-O-Zn和O-Zn-O的健角比纯净的要小,呈现出压缩状态。对于model
2,Ag掺杂在第二层时的Zn-O健长大约为1.864 Å,和纯净中的Zn-O健要相差不大。而Zn-O-Zn的健角减小得比较多。对于model 3,Zn-O健长为1.861
Å,O-Zn-O健角为105.006˚,Zn-O-Zn健角为113.003˚。其中健长变短,而O-Zn-O健

Figure 1. The models of Ag incorporation at different
sites on ZnO(10 0) surface .
0) surface .
图1. Ag嵌入在ZnO(10 0)表面的不同位置的模型
0)表面的不同位置的模型

Table 1. Calculation structural parameters of various models
表1. 各种模型的计算的结构参数

Figure 2. The relaxation structure of Ag incorporation
at different site on ZnO(10 0) surface
0) surface
图2. Ag嵌入ZnO(10 0)表面不同位置的弛豫结构图
0)表面不同位置的弛豫结构图
角变大,Zn-O-Zn健角变小。而对于model 4,和纯净ZnO(10 0)表面相比,Zn-O健的健长相对来说变长,健角都变小。
0)表面相比,Zn-O健的健长相对来说变长,健角都变小。
从以上分析以及图2中我们发现:Ag嵌入到ZnO(10 0)面的表面层时对晶格畸变影响较大,而嵌入到中间层时晶格畸变影响较小。这个弛豫发生明显的变化主要归因于量子杂化效应和库仑作用。在形成ZnO(10
0)面的表面层时对晶格畸变影响较大,而嵌入到中间层时晶格畸变影响较小。这个弛豫发生明显的变化主要归因于量子杂化效应和库仑作用。在形成ZnO(10 0)表面时,在表面处的Zn和O都构成悬挂键。由于能量遵循能量最低的原则,所以表面的悬挂键进行重新组合。从而,表面氧原子轨道杂化类型为p3,导致氧原子向体外移动;而Zn原子杂化类型为sp2,导致Zn原子向体内移动。此外,由于表面的Zn原子和O原子的电负性的差别很大,从而使得在表面发生电子的转移,Zn原子的悬挂键电子向O原子转移,导致两原子之间的库能作用增强,使得表面发生收缩,从而发生很大扭曲。
0)表面时,在表面处的Zn和O都构成悬挂键。由于能量遵循能量最低的原则,所以表面的悬挂键进行重新组合。从而,表面氧原子轨道杂化类型为p3,导致氧原子向体外移动;而Zn原子杂化类型为sp2,导致Zn原子向体内移动。此外,由于表面的Zn原子和O原子的电负性的差别很大,从而使得在表面发生电子的转移,Zn原子的悬挂键电子向O原子转移,导致两原子之间的库能作用增强,使得表面发生收缩,从而发生很大扭曲。
3.2. 功函数
图3为Ag嵌入ZnO(10 0)表面不同位置时的静电势。从图中,我们发现,随着Ag嵌入到近表面层,静电势具有独特的特性,并且在model
1中有很大的变化,这主要是由于表面弛豫引起的。同时,我们发现静电势沿着表面向下弯曲,这表明Ag原子嵌入之后,有一定的内部电荷向表面转移。功函数的计算公式为:
0)表面不同位置时的静电势。从图中,我们发现,随着Ag嵌入到近表面层,静电势具有独特的特性,并且在model
1中有很大的变化,这主要是由于表面弛豫引起的。同时,我们发现静电势沿着表面向下弯曲,这表明Ag原子嵌入之后,有一定的内部电荷向表面转移。功函数的计算公式为:
 (1)
(1)
其中,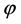 为功函数,
为功函数,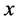 是真空静电势,
是真空静电势,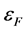 为费米能级。如表2所示,我们计算所得的纯净ZnO(10
为费米能级。如表2所示,我们计算所得的纯净ZnO(10 0)面的表面功函数为5.350 eV。这与实验值为5.300
eV[12] 相吻合。
0)面的表面功函数为5.350 eV。这与实验值为5.300
eV[12] 相吻合。
对于Ag掺杂ZnO表面的不同位置的功函数如表2所示。从这里我们发现,纯净ZnO表面的功函数比Ag掺杂ZnO表面的不同位置的功函数小,而且当Ag嵌入在第一层时,功函数值最大,这说明Ag原
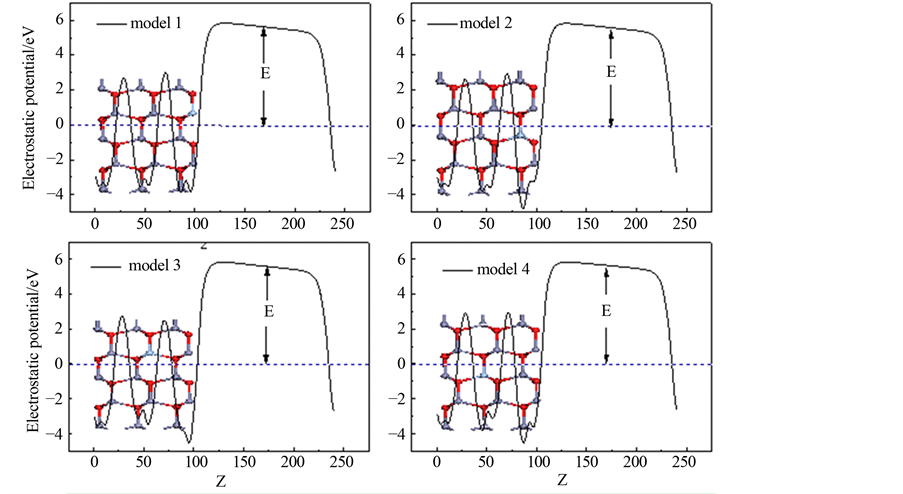
Figure 3. The electrostatic potentials of Ag incorporation
on ZnO(10 0) surface
0) surface
图3. Ag在ZnO(10 0)表面嵌入不同位置的静电势
0)表面嵌入不同位置的静电势
Table 2. The formation and ionization energy,
and work function of Ag incorporation on ZnO(10 0) surface
0) surface
表2. Ag嵌入ZnO(10 0)表面的形成能、离化能和功函数
0)表面的形成能、离化能和功函数
子嵌入第一层时,电子很容易迁移到表面。
3.3. 形成能和离化能
为了进一步了解Ag嵌入ZnO(10 0)表面的导电机制,我们计算了Ag在ZnO(10
0)表面的导电机制,我们计算了Ag在ZnO(10 0)表面上的不同位置的形成能。各种模型的形成能可以定义[13] 为:
0)表面上的不同位置的形成能。各种模型的形成能可以定义[13] 为:
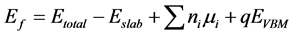 (2)
(2)
其中 为含缺陷的模型的总能;
为含缺陷的模型的总能; 为纯净ZnO(10
为纯净ZnO(10 0)表面的总能;
0)表面的总能;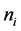 是i元素的个数,当i加进体系时,
是i元素的个数,当i加进体系时,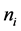 为负;当替换时,
为负;当替换时, 为正;
为正;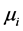 是i的化学势;
是i的化学势;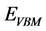 为纯净ZnO相对于价带顶的能量;为了保持稳定的ZnO表面,避免其他复合体的生成,应该满足以下关系式:
为纯净ZnO相对于价带顶的能量;为了保持稳定的ZnO表面,避免其他复合体的生成,应该满足以下关系式:
 (3)
(3)
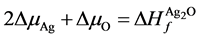 (4)
(4)
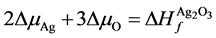 (5)
(5)
其中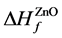 ,
,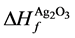 ,
, 分别为ZnO,Ag2O3以及Ag2O的形成熵。
分别为ZnO,Ag2O3以及Ag2O的形成熵。
离化能为当缺陷D带电荷q时的形成能和当缺陷D带电荷 时的形成能相等时的费米能级就是离化能。因此,离化能可以这样计算[14]
[15] 。
时的形成能相等时的费米能级就是离化能。因此,离化能可以这样计算[14]
[15] 。
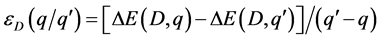 (6)
(6)
Ag嵌入ZnO(10 0)表面的不同位置的形成能如表2所示。我们发现Ag嵌入ZnO(10
0)表面的不同位置的形成能如表2所示。我们发现Ag嵌入ZnO(10 0)表面时处于第一层时的形成能小于掺杂在其他层的情况,这表明嵌入在表面时的结构稳定,而且Ag原子很容易掺杂进去。比较Ag嵌入ZnO(10
0)表面时处于第一层时的形成能小于掺杂在其他层的情况,这表明嵌入在表面时的结构稳定,而且Ag原子很容易掺杂进去。比较Ag嵌入ZnO(10 0)表面的几种情况发现,从外层逐渐向内部时相应的形成能逐渐增加,并且逐渐接近于Ag掺杂块体ZnO的形成能。因此,我们可以得知:Ag原子很容易占据Zn位并且掺到ZnO(10
0)表面的几种情况发现,从外层逐渐向内部时相应的形成能逐渐增加,并且逐渐接近于Ag掺杂块体ZnO的形成能。因此,我们可以得知:Ag原子很容易占据Zn位并且掺到ZnO(10 0)的近表面,并且在表面形成稳定结构。接着,我们计算了嵌入体系的离化能。从表2中发现Ag嵌入ZnO(10
0)的近表面,并且在表面形成稳定结构。接着,我们计算了嵌入体系的离化能。从表2中发现Ag嵌入ZnO(10 0)第一层的离化能比掺杂在第三层的离化能高,而比掺杂在第二层的离化能高很多。同时,我们发现嵌入在ZnO(10
0)第一层的离化能比掺杂在第三层的离化能高,而比掺杂在第二层的离化能高很多。同时,我们发现嵌入在ZnO(10 0)表面第四层的离化能和Ag掺杂ZnO块体材料的离化能相近。因此,我们得出结论:Ag嵌入ZnO(10
0)表面第四层的离化能和Ag掺杂ZnO块体材料的离化能相近。因此,我们得出结论:Ag嵌入ZnO(10 0)表面很容易处于表面层,但是离化能比较高,不易离化,这样阻碍了形成较稳定的p型导电材料。
0)表面很容易处于表面层,但是离化能比较高,不易离化,这样阻碍了形成较稳定的p型导电材料。
3.4. 电子结构
图4给出了Ag嵌入ZnO(10 0)表面以及纯净ZnO(10
0)表面以及纯净ZnO(10 0)表面的态密度图,其中虚线为费米能级(
0)表面的态密度图,其中虚线为费米能级(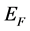 )。通过前面的研究[16] 发现,纯净ZnO(10
)。通过前面的研究[16] 发现,纯净ZnO(10 0)表面的价带顶主要有O-2p态电子决定,而导带低主要由Zn-4s和O-2p共同作用。同时,我们研究组[16]
通过研究也发现:ZnO(10
0)表面的价带顶主要有O-2p态电子决定,而导带低主要由Zn-4s和O-2p共同作用。同时,我们研究组[16]
通过研究也发现:ZnO(10 0)表面的态密度图反应出了明显的表面态。其中,O-2p态电子逐渐向价带顶区域移动,在表面构成成键态的表面态;而Zn-4s态电子向导带底区域移动,在表面构成了反键态的表面态,因而,由于价带顶向高能方向移动与导带底向低能方向移动的共同作用,使得ZnO(10
0)表面的态密度图反应出了明显的表面态。其中,O-2p态电子逐渐向价带顶区域移动,在表面构成成键态的表面态;而Zn-4s态电子向导带底区域移动,在表面构成了反键态的表面态,因而,由于价带顶向高能方向移动与导带底向低能方向移动的共同作用,使得ZnO(10 0)表面的带隙变窄,这也是表面材料与块体材料态密度主要的区别。然而,和纯净ZnO(10
0)表面的带隙变窄,这也是表面材料与块体材料态密度主要的区别。然而,和纯净ZnO(10 0)表面相比(如图4所示),随着Ag嵌入到ZnO(10
0)表面相比(如图4所示),随着Ag嵌入到ZnO(10 0)表面,费米能级逐渐由本征态移向p型状态转移。从这里得知,Ag嵌入ZnO(10
0)表面,费米能级逐渐由本征态移向p型状态转移。从这里得知,Ag嵌入ZnO(10 0)表面表现为p型,然而由于表面效应的存在使得表
0)表面表现为p型,然而由于表面效应的存在使得表
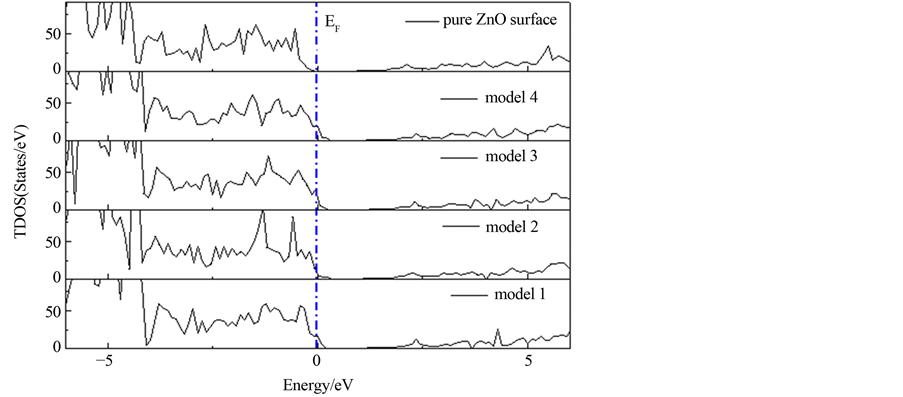
Figure 4. The density of states of Ag incorporation
on ZnO(10 0) surface
0) surface
图4. Ag在ZnO(10 0)表面嵌入不同位置的态密度图
0)表面嵌入不同位置的态密度图
面的Ag原子离化能较大,这样阻碍了制备p型ZnO材料。
4. 结论
本文通过第一性原理计算方法研究了纯净ZnO(10 0)及Ag嵌入ZnO(10
0)及Ag嵌入ZnO(10 0)表面的晶格结构和导电性以及电子结构。通过和纯净ZnO(10
0)表面的晶格结构和导电性以及电子结构。通过和纯净ZnO(10 0)表面结构相比较,发现,Ag嵌入ZnO(10
0)表面结构相比较,发现,Ag嵌入ZnO(10 0)使其结构发生较大变化。同时,通过计算Ag原子嵌入ZnO(10
0)使其结构发生较大变化。同时,通过计算Ag原子嵌入ZnO(10 0)表面的形成能,发现Ag原子在ZnO(10
0)表面的形成能,发现Ag原子在ZnO(10 0)近表面位置最为稳定,Ag原子处于表面层。而在中间原子的形成能接近于块体材料掺杂的形成能,但处于表面的Ag原子不易离化,导致制备p型ZnO的困难。通过本文的研究为实验上Ag掺杂ZnO薄膜材料提供有利的理论支持。
0)近表面位置最为稳定,Ag原子处于表面层。而在中间原子的形成能接近于块体材料掺杂的形成能,但处于表面的Ag原子不易离化,导致制备p型ZnO的困难。通过本文的研究为实验上Ag掺杂ZnO薄膜材料提供有利的理论支持。
参考文献 (References)
- [1] Zeng, Y.J., Ye, Z.Z., Xu, W.Z., Li, D.Y., Lu, J.G., Zhu, L.P. and Zhao, B.H. (2006) Dopant source choice for formation of p-type ZnO: Li acceptor. Applied Physics Letters, 88, Article ID: 062107.
- [2] Wan, Q.X., Xiong, Z.H., Dai, J.N., Rao, J.P. and Jiang, F.Y. (2008) First-principles study of Ag-based p-type doping difficulty in ZnO. Optical Materials, 30, 817-821.
- [3] Xu, W.Z., Ye, Z.Z., Zhou, T., Zhao, B.H., Huang, J.Y., et al. (2004) Low-pressure MOCVD growth of p-type ZnO thin films by using NO as the dopant source. Journal of Crystal Growth, 265, 133.
- [4] Tae, H.K, Jin, J.P. and Sang, H.N, (2009) Fabrication of Mg-doped ZnO thin films by laser ablation of Zn: Mg target. Applied Surface Science, 255, 5264-5266.
- [5] Fan, J. and Freer, R. (1995) The roles played by Ag and Al dopants in controlling the electrical properties of ZnO varistors. Journal of Applied Physics, 77, 4795-4800.
- [6] Gouvea, C.A.K., Wypych, F. and Moraes, S.G. (2000) Semiconductor-assisted Photodegradation of lignin, dye, and Kraft effluent by Ag-doped ZnO. Chemosphere, 40, 427-432.
- [7] Xue, H., Xu, X.L., Chen, Y., et al. (2008) Influence of Ag-doping on the optical properties of ZnO films. Applied Surface Science, 255, 1806-1810.
- [8] Duan, L., Lin, B.X. and Fu, Z.X. (2006) Enhancement of ultraviolet emissions from ZnO Films by Ag doping. Applied Physics Letters, 88, 232110-1-232110-232113.
- [9] Charton, C. and Fahland, M. (2003) Optical properties of thin Ag films deposited by magnetron sputtering. Surface and Coatings Technology, 174-175, 181-186.
- [10] Duan, L., Lin, B.X. and Fu, Z.X. (2006) Enhancement of ultraviolet emissions from ZnO films by Ag doping. Applied Surface Science, 88, 232110-1-232110-232113.
- [11] Kresse, G. and Hafner, J. (1994) Ab initio molecular dynamics for liquid metals. Physical Review B, 47, 558.
- [12] Kresse, G. and Furthermuller, J. (1996) Efficiency of ab-initio total energy calculations using a plane-wave basis set. Physical Review B, 54, 11169.
- [13] Lee, C.J., Lee, T.J., Lyu, S.C., Zhang, Y., Ruh, H. and Lee, H.J. (2002) Field emission from well aligned zinc oxide nanowires grown at low temperature. Applied Physics Letters, 81, 3648(1-3).
- [14] Xu, H., Zhang, R.Q. and Tong, S.Y. (2010) Interaction of O2,
H2O, N2 and O3 with stoichiometric and reduced
ZnO (10
 0) surface. Physical Review B, 82, 155326(1-6).
0) surface. Physical Review B, 82, 155326(1-6). - [15] Yan, Y.F. and Wei, S.H. (2008) Doping asymmetry in wide-bandgap semiconductors: Origins and solutions. Physica Status Solidi (b), 245, 641-652.
- [16] 熊志华 (2008) ZnO掺杂改性的第一性原理研究. 博士学位论文, 南昌大学, 南昌.