Hans Journal of Computational Biology
Vol.
09
No.
01
(
2019
), Article ID:
29394
,
7
pages
10.12677/HJCB.2019.91001
Reconstruction of Single-Cell Chromosome Three-Dimensional Structure Based on Surface Fitting
Liwei Liu, Qi Zhang, Fenglan Bai
College of Science, Dalian Jiaotong University, Dalian Liaoning

Received: Mar. 4th, 2019; accepted: Mar. 18th, 2019; published: Mar. 25th, 2019

ABSTRACT
Genomics is one of the core areas of bioinformatics; there are two main research directions of genomics, structural genomics targeting whole genome sequencing and functional genomics targeting gene function interpretation. In the past few decades, genomics has experienced considerable development. The prediction of the three-dimensional structure of chromosomes is of great significance for the study of genomics. The reconstruction of the three-dimensional structure of chromosomes is to predict the conformation of the three-dimensional image from the one-dimensional and two-dimensional data of the genome, and then use the data analysis method to judge the reliability of the three-dimensional structure of the reconstructed chromosome. This paper is based on the single-cell chromosome Hi-C technology and Hi-3C derived data to capture the interaction data of individual cells, write the contact frequency matrix, and then convert the contact frequency matrix into a distance matrix to further obtain the three-dimensional structure of the chromosome. The contact matrix of single-cell Hi-C data is sparse and noise-containing, missing many non-contact sites. We refer to such a matrix as a low-rank matrix. The first problem we have to solve is the processing of low rank matrices, also called the completion of low rank distance matrices. This paper introduces several common low-rank matrix completion methods including optimization method and shortest distance method. It also introduces the different methods used in this paper. Finally, the final conclusion is obtained through MATLAB and compared of human research results.
Keywords:Hi-C Data, Low-Rank Matrix, Completion of Low-Rank Matrices, Shortest Distance Method
基于曲面拟合重建单细胞染色体三维结构
刘立伟,张琦,白凤兰
大连交通大学理学院,辽宁 大连
收稿日期:2019年3月4日;录用日期:2019年3月18日;发布日期:2019年3月25日

摘 要
基因组学是当今生物信息学的核心领域之一,基因组学的两个主要研究方向是:以全基因组测序为目标的结构基因组学和以基因功能解读为目标的功能基因组学。在过去几十年,基因组学经历了长足的发展。而染色体三维结构的预测对基因组学的研究有重大意义。染色体三维结构的重建问题,就是从基因组的一维和二维数据出发预测其在三维空间中的构像,再利用数据分析等方法判断重建后染色体三维结构的可靠性。单细胞的Hi-C数据的接触矩阵是稀疏且含有噪声的,缺失很多接触位点的信息,我们把这样的矩阵称作低秩矩阵。我们首先要解决的问题就是对于低秩矩阵的处理,也叫低秩距离矩阵的完备化。本文介绍了包括最优化方法、最短距离法在内的几种常见的低秩矩阵完备化的方法,也详细介绍了本文采用的与前人不同的方法,最后通过MATLAB实现得到最终结论并与前人研究成果形成对比。
关键词 :Hi-C数据,低秩矩阵,低秩矩阵的完备化,最短距离法

Copyright © 2019 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 引言
在一个经典的染色体构象捕获技术(chromosome conformation capture, 3C)实验中,要处理千千万万个细胞中的染色体交互信息,首先,利用甲醛交联染色体,目的在于固定蛋白质和DNA,使染色体具有相对稳定的三维结构;然后,用限制性内切酶分解,这些交叉结合的碎片结扎形成混合DNA分子。混合DNA分子包括被捕获的相互作用的各个方面。在3C实验中,检测结扎产品是通过PCR方法利用特定位点引物 [1];4C实验中利用PCR反方向产生单位点基因组范围的相互作用剖面图;5C是3C技术与热处理和结扎寡核苷酸混合交互的方法;Hi-C技术是无偏性和3C基因组范围适应的首个技术 [1],这个方法包括了一步将生物素华的核苷酸作为酶切位点,便于隔离结扎产品和增加后序的测序工效。
除了以上基于3C基础上的技术外,光学和电子显微镜可以提供基因层次上的结构观察。虽然光学显微镜受到光衍射的性质,使物体接近200纳米的时候很难分离,这里有一个新的技术使这个阈值降低到10~20纳米。这个方法是基于通过改变激发光源来取样的调查基础上。除了高分辨率的光学显微镜,低分辨率的显微镜很难分清染色质纤维的折叠和压实的情况。电子显微镜可以观察到染色质纤维的折叠和压实的情况,但是,电子显微镜容易损坏,且无法观察活细胞。
3C实验中产生的接触信息是一个低秩矩阵,染色体的接触频率矩阵是一个0-1矩阵,有接触记为1,无接触记为0。然而0的数目居多,这导致这个接触频率矩阵的秩很低,在转换大距离矩阵时会丢失很多数据。把上述矩阵叫做低秩矩阵。目前,低秩矩阵的完备化一般有两种思路:从优化角度完备低秩矩阵和从数值分析角度完备低秩矩阵。下面分别介绍这两种思路。
2. 常见的缺失数据推断方法
2.1. 利用最优化方法推断缺失数据
给出一组n个不同数据点的欧式距离矩阵 ,距离矩阵的完备化理论是要解决以下问题 [2] :
(1)
其中,H是对称的二进制矩阵,算子
表示广义的矩阵乘积,如果D中给定的有序数组
,满足
,则
D中元素个数用d表示,显然d最多等于 ,在大多数应用中,它的秩为 ,其中r是最佳嵌入维数,这种度量距离并不是真正意义上的距离,不满足三角不等式。
对于(1)有一个有效的可供选择的想法,那就是将问题(1)转化为半正定矩阵的最优化问题 [2] 。这个想法来自于Schoenberg的一个经典结论——欧式距离矩阵的秩和半正定矩阵的秩等于嵌入空间的维数 [2] 。则(1)式就可以写成
(2)
其中,k是半正定矩阵与欧式距离矩阵的转化关系:
, 定义为提取对角线元素,1表示所有和1相等的矢量 [2] 。
(2)比(1)有一个实用性的好处,那就是X的秩等于嵌入空间的维数,当矩阵X的秩无约束时,问题(2)是凸的,因此能够解决问题 [2] 。
在这篇文章中,我们认为问题(2)的解 是低秩的,即
 (3)
(3)
在非凸问题中,维数一直增加到r的真实值,每一个非凸问题都存在一个被控制秩的最优化问题 [3],即
使得 (4)
通过把p值从1取到r,参考文献 [2] 中给出的结果保证了问题(2)结果的收敛性。问题(4)的有效解决取决于找到一个低秩参数,因为任意一个秩为p的半正定矩阵X都可以因式分解为:
,其中 为了找到这个矩阵分解,我们采用黎曼流形中的几何框架最优化。参考文献 [3] [4] 介绍了矩阵流形最优化更多细节和最新研究成果。
综上所述:低秩矩阵完备化的最优化方法就上将低秩矩阵转化为一个半正定矩阵的优化问题 [5] [6] 。
2.2. 利用最短距离法推断缺失数据
Lieberman-Aiden等人研究了两个片段的接触频率、基因线性距离和空间距离之间的关系,表明空间上越接近的片段接触频率值越大,空间距离远的片段产生的接触频率值越小。因此在ShRec3D中有了(5)式的转换函数。Floyd-Warshall算法是一种经典的最短路径算法 [2],可以求解图中任意两点之间的最短路径。其主要过程如下:
1) 构建加权图 [7] 。假设一个二元组 表示一个无向图,其中V表示非空顶点集,每个染色体片段表示图中的顶点;E是无序积V&V的一个多重子集,称E为G的边集,有接触的任意两个片段之间存在一条边,组成边集E [2] 。
2) Floyd-Warshall算法步骤。①令
,输入权重矩阵
。②令
,计算
,式中
。③如果k = n,终止算法;否则,返回步骤②。经过这三步,最终结果
中元素
就是从顶点
到 的最短路径,从而获得真
的最短路径,从而获得真
正意义上的距离矩阵D。Floyd算法是一种动态规划算法,可以求取无向加权图中任意两点之间的最短路径距离,但是要求Hi-C数据构建的加权图是连通的 [2] 。Hi-C技术捕获的是整个细胞系中数百万个细胞的染色体片段的接触信息,所以由染色体片段作为顶点构建的权重图理论上是连通的,这也是最短路径算法应用的前提 [2] 。
3. 利用Hi-C数据得到染色体三维结构
此过程主要是分成以下三步。
3.1. 将单细胞Hi-C接触矩阵转化为空间距离矩阵
 (5)
(5)
其中有交互作用的片段之间的空间距离为1,其余的无接触的空间距离为0 [5] 。经过(5)式的转化,我们就能获得单细胞染色体片段间的接触频率矩阵,该矩阵是稀疏的。
3.2. 通过接触频率矩阵得到欧氏距离矩阵
在科学界普遍认为,片段间接触频率与欧氏距离呈负相关。这点很容易理解:距离越远,越不容易接触,导致接触频率越低 [8] 。由于接触频率矩阵中零元素很多,导致矩阵噪声过大,在转化过程中不好处理,所以我们先利用交并运算,补出一部分缺失数据 [9] [10],具体计算公式如下:
(6)
仅公式(6)推断出来的缺失数据还是少数。然后我们利用插值函数将所有零值全部推断出,这样得到的矩阵中的元素有正有负,我们将所有元素都映射到[0, 1]区间,公式如下:
(7)
其中 是插值之后的矩阵中的元素, 是所有矩阵元素的最大值, 是所有矩阵元素的最
小值,通过该映射,接触频率矩阵中的元素就都与[0, 1]区间中的元素一一对应,而且各元素之间的相对大小关系还不发生改变。由于接触频率自身的性质,决定了接触频率矩阵对角线元素都为1,也就是片段与自身肯定有接触!
经过以上处理,我们得到了最终的接触频率矩阵,它具有以下特点:
1)主对角线上元素为1;
2)每个元素都在[0, 1]区间内;
接着,我们将接触频率矩阵转化为距离矩阵,我们设上一步得到的频率矩阵为F。
转化公式如下:
(8)
这样,我们就得到了欧式距离矩阵D。
3.3. 利用MDS方法得到染色体三维坐标
MDS [2] 算法的主要步骤如下:
1) 定义一个度量矩阵M,其中M可用距离矩阵D通过(6)式计算得到。
(9)
其中 为染色体第i个片段和第j个片段之间的距离,M为对称矩阵。
2) 将M矩阵进行下式的特征值分解。
(10)
其中A是对角矩阵,其元素是矩阵M分解所得的特征值,V是A的对角特征值对应的特征向量按照列排列组成的矩阵,则其k维坐标可由下式计算得到 [11] 。
(11)
本文求染色体片段的三维坐标,则 保留最大的三个特征值和对应的特征向量。假设染色体片段
的三维坐标为 ,矩阵M的最大的三个特征值为 ,对应的特征向量为 [2] [12] 。则第i个片段的三维坐标可由下式计算得出:
(12)
经MDS的维数约减计算,可以获得三维空间中染色体片段的空间坐标,用可视化软件可以生动的呈现染色体3D结构,进行生物意义的探索算法性能分析 [2] 。
4. 结论
4.1. 阐述结论
根据我们的方法,利用MATLAB画出染色体三维结构,如图1,图2所示:
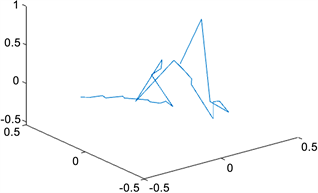
Figure 1. Structure of the X chromosome at resolution 6,000,000
图1. X染色体在分辨率为6,000,000时的结构

Figure 2. Structure of the X chromosome at resolution 8,000,000
图2. X染色体在分辨率为8,000,000时的结构
4.2. 比较
首先看热图的不同,用我们的方法画出来的热图(见图3)对比于Jonas Paulsen等人应用MBO算法的热图,接触信息更多,如图所示,利用方法MBO得到的热图只能得到主对角线附近接触位点的接触信息,数据点太少。
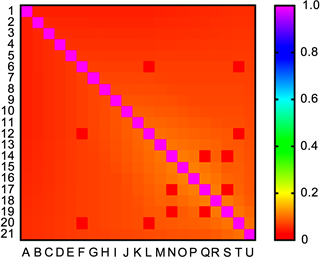
Figure 3. Heat map of the contact frequency of each contact site on the X chromosome at a resolution of 8,000,000
图3. X染色体在分辨率为8,000,000时每个接触位点接触频率的热图
对比于Jonas Paulsen等人应用MBO算法 [2],借助Matlab中的优化工具箱Manopt重构的染色体结构(见图4),我们采取的方法有以下特点:

Figure 4. Jonas Paulsen uses the optimization toolbox in Matlab to reconstruct the chromosome structure of Manopt
图4. Jonas Paulsen 用Matlab中的优化工具箱Manopt重构的染色体结构
1) 得到了更多的接触频率,使得重构的染色体结构给出染色体非主体部位的结构,弱化了接触位点对于重构后结构的影响,着重描写了染色体的整体结构,即用简单的线条连接每一个接触位点,使每个接触位点都很渺小,从而不影响整体的重构结构;
2) 补充Jonas Paulsen等人的结果中重合部分的结构,如下图所示,Jonas Paulsen等人重构的X染色体3D结构中间部位全是各个接触位点重合的现象,根本没有给出具体的染色体3D结构。这样更有利于人们更好地把握染色体的整体结构。
致谢
本文得到中国博士后科学基金项目(编号2018M631782),辽宁省自然科学基金项目(编号201800278),经费5万;辽宁省教育厅项目(编号L2015093)资助。
文章引用
刘立伟,张 琦,白凤兰. 基于曲面拟合重建单细胞染色体三维结构
Reconstruction of Single-Cell Chromosome[J]. 计算生物学, 2019, 09(01): 1-7. https://doi.org/10.12677/HJCB.2019.91001
参考文献
- 1. 彭城, 李国亮, 张红雨, 等. 染色质三维结构重建及其生物学意义[J]. 中国科学: 生命科学, 2014, 44(8): 794-802.
- 2. 张卫. 基于Hi-C数据的预测染色体三维结构的方法研究[D]: [硕士学位论文]. 北京: 北京工业大学, 2016.
- 3. Paulsen, J., Gramstad, O. and Collas, P. (2015) Mainifold Based Optimization for Single-Cell 3D Ge-nome Reconstruction. PLoS Computational Biology, 11, e1004396. https://doi.org/10.1371/journal.pcbi.1004396
- 4. 李建更, 张卫, 李晓丹. 基于参数优化的染色体三维结构预测算法VMBO [J]. 北京工业大学学报, 2018, 44(2): 207-214.
- 5. Mishra, B., Meyer, G. and Sepulchre, R. (2011) Low-Rank Optimization for Distance Matrix Completion. 2011 50th IEEE Conference on Decision and Control and European Control Conference, Orlando, FL, 12-15 December 2011, 4455-4460. https://doi.org/10.1109/CDC.2011.6160810
- 6. Mishra, B. (2014) A Riemannian Geometry for Low-Rank Matrix Completion.
- 7. 项荣武, 刘艳杰, 胡忠盛. 图论中最短路径问题的解法[J]. 沈阳航空工业学院学报, 21(2): 86-88.
- 8. Hirata, Y., Oda, A., Ohta, K. and Aihara, K. (2016) Three-Dimensional Reconstruction of Single-Cell Chromosome Structure Using Recurrence Plots. Scientific Reports, 6, Article No. 34982. https://doi.org/10.1038/srep34982
- 9. Hirata, Y., Horai, S. and Aihara, K. (2008) Reproduction of Distance Ma-trices and Original Time Series from Recurrence Plot and Their Applications. The European Physical Journal Special Topics, 164, 13-22. https://doi.org/10.1140/epjst/e2008-00830-8
- 10. Tanio, M., Hirata, Y. and Suzuki, H. (2009) Reconstruction of Driving Forces through Recurrence Plots. Physics Letters A, 373, 2031-2040. https://doi.org/10.1016/j.physleta.2009.03.069
- 11. Varoquaux, N., Ay, F., Noble, W.S. and Vert, J.P. (2014) A Statistical Approach for Inferring the 3D Structure of the Genome. Bioinformatics, 30, i26-i33. https://doi.org/10.1093/bioinformatics/btu268
- 12. Ben-Elazar, S., Yakhini, Z. and Yanai, I. (2013) Spatial Lo-calization of Co-Regulated Genes Exceeds Genomic Gene Clustering in the Saccharomyces cerevisiae Genome. Nucleic Acids Research, 41, 2191-2201. https://doi.org/10.1093/nar/gks1360