Hans Journal of Computational Biology
Vol.
11
No.
04
(
2021
), Article ID:
53129
,
10
pages
10.12677/HJCB.2021.114006
基于融合特征和Voting集成学习的 膜蛋白类型预测
苏鹏程
浙江理工大学,浙江 杭州
收稿日期:2022年5月21日;录用日期:2022年6月21日;发布日期:2022年6月30日

摘要
膜蛋白是细胞功能的主要承担者,其功能与其类型密切相关。膜蛋白类型的鉴定是生物信息学中的一项重要课题。已有的膜蛋白分类模型主要从膜蛋白序列信息中提取特征,本文提出了一种基于蛋白质二级结构信息的蛋白质特征提取方法,并将其融入现有的两种序列特征。通过对比实验结果显示,在融入了蛋白质二级结构特征后,几种不同机器学习分类算法下的膜蛋白预测精度均有提升,说明了该融合蛋白质二级结构特征方法的有效性。最后,基于Voting集成学习框架,结合三种机器学习算法构建膜蛋白分类模型。结果表明,该模型的预测效果优于现有的几种机器学习模型。
关键词
膜蛋白分类,蛋白质二结构,特征融合,机器学习,Voting集成学习

Prediction of Membrane Protein Types Based on Fusion Feature Information and Voting Ensemble Learning
Pengcheng Su
Zhejiang Sci-Tech University, Hangzhou Zhejiang
Received: May 21st, 2022; accepted: Jun. 21st, 2022; published: Jun. 30th, 2022

ABSTRACT
Studies have shown that membrane proteins are the main bearers of cellular functions and their functions are closely related to their types. Therefore, the identification of membrane protein types is an important topic in bioinformatics. The existing classification models for membrane proteins mainly extract features from the sequence information of membrane proteins. In this paper, a protein feature extraction method was proposed based on protein secondary structure information, which was integrated into two existing sequence features. By comparing the experimental results, the prediction accuracy of membrane proteins under several different machine learning classification algorithms was improved after integrating protein secondary structure features, which illustrated the effectiveness of this fusion protein secondary structure feature method. Finally, a membrane protein classification model was constructed based on the voting ensemble learning framework in combination with three machine learning algorithms. The results show that the prediction performance of this model is better than other machine learning models.
Keywords:Classification of Membrane Protein, Protein Secondary Structure, Feature Fusion, Machine Learning, Voting Ensemble Learning

Copyright © 2021 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/


1. 引言
蛋白质是生命的物质基础,膜蛋白作为一种重要的蛋白类型,具有许多重要的功能,它们参与许多重要的细胞反应。包括作为载体将物质运入和运出细胞,作为蛋白激素的特异性受体,执行细胞识别功能,负责信号传递和细胞间的相互作用等 [1]。膜蛋白与药物研发密切相关,目前,已经证实60%以上的药物靶向目标都是膜蛋白 [2]。因此,膜蛋白的研究一直是生物信息学中的重要课题。
膜蛋白的功能与其类型密切相关,传统的生物实验是确定膜蛋白类型最可靠的方法,但是它效率低且成本高。因此,设计一种速度快、成本低、精度高的膜蛋白类型鉴定的计算方法是一项迫切的任务,机器学习方法就是最常用的方法之一。
根据膜蛋白与脂质双分子层的相互作用关系,膜蛋白可分为八类:I型单跨膜蛋白、II型单跨膜蛋白、III型单跨膜蛋白、IV型单跨膜蛋白、外周膜蛋白、多跨膜蛋白、GPI锚膜蛋白和脂质链锚膜蛋白 [3]。
对于机器学习模型来说,特征提取是关键的一步。Chou等人首先提出用氨基酸组成(amino acid composition, AAC)来表示膜蛋白 [4]。但是,由于AAC只包含氨基酸的成分信息,Chou等人进一步提出伪氨基酸组成(pseudo amino acid composition, PseAAC)来解决这个问题 [5]。Hayat等人使用拆分氨基酸组成(split amino acid composition, SAAC)来表示膜蛋白 [6]。
此外,从蛋白质序列中提取的其它特征也被用于膜蛋白分类,如二肽组成(dipeptide composition, DipC) [7],四肽组成(tetra peptide pattern, TPP) [8],位置特异性得分矩阵(position specific scoringmatrix, PSSM)等。
另一方面,许多机器学习算法被用于构建膜蛋白分类模型。例如,Hayat等人提出了一个随机森林(RF) [9] 模型来对膜蛋白类型进行分类。类似地,支持向量机(SVM) [10]、K近邻(KNN) [11]、朴素贝叶斯(NBM) [12] 和极端随机树 [13] 也被用来构造分类模型。
已有的膜蛋白分类模型主要从蛋白质一级结构(序列)中提取特征,蛋白质的结构决定其功能 [14],从蛋白质结构中提取的蛋白质信息与从蛋白质序列中提取的蛋白质信息有很大的不同,因此蛋白质的结构信息可以用于膜蛋白分类。本文提出了一种融合蛋白质二级结构信息和蛋白质序列信息的膜蛋白特征提取方法,并通过对比实验证明了该特征提取方法的有效性。最后基于此融合特征模型与Voting集成学习框架构建了一个膜蛋白分类模型。结果表明,本文提出的模型优于现有的几种机器学习模型。
2. 材料与方法
2.1. 数据集
本文使用的数据集是Wan等人建立的。该数据集共包含7582条膜蛋白数据,其中训练集包含3249条膜蛋白数据,测试集包含4333条膜蛋白数据 [15]。各类型样本的详细分布情况如表1所示。

Table 1. Distribution of sample types across membrane protein datasets
表1. 膜蛋白数据集的各类型样本分布表
2.2. 特征提取
为了构建高效的分类模型,特征提取是一个关键步骤。在膜蛋白分类预测中,一个好的特征提取方法可以从原始数据中提取尽可能多的隐藏信息。为了获得一个好的特征模型,本文从蛋白质二级结构信息中提取特征,并将其与基于蛋白质序列的特征融合起来表示蛋白质。
2.2.1. 拆分氨基酸组成(SAAC)
AAC模型使用蛋白质中20种氨基酸出现的频率表示蛋白质。在SAAC模型中,每个膜蛋白序列被拆分为3个片段,(1) 25个N端氨基酸,(2) 25个C端氨基酸,以及(3)这两个端之间的片段。分别对3个蛋白质片段计算AAC得到3个20维的特征向量,之后将得到的3个AAC向量拼接成一个特征向量。最终,一个膜蛋白序列被转化成一个60维的特征向量。相较于AAC模型,SAAC模型包含了部分氨基酸的顺序信息。
2.2.2. 伪氨基酸组成(PseAAC)
PseAAC模型不仅包含AAC的信息,还加入了部分氨基酸排列顺序信息和理化性质信息(如氨基酸的疏水性、亲水性、侧链质量等)。
给定长为L的蛋白质 ,其氨基酸顺序信息和理化性质信息使用下面的顺序相关因子表示:
(1)
其中: 被称为第i层顺序因子,它表示蛋白质序列上所有间隔为i的氨基酸残基之间的顺序相关因子(如图1所示)。相关函数 的定义如下:
(2)
其中:n表示所使用氨基酸理化性质的种类数, 表示蛋白质序列中第i个氨基酸残基的第k种理化性质的值。在将各种理化性质的值代入式子之前,都需要使用公式进行标准化。
(3)
其中: 表示蛋白质序列中,第i个氨基酸残基的第k种理化性质的原始值。

Figure 1. Layer i sequence order correlation factors for protein sequences
图1. 蛋白质序列的第i层序列顺序相关因子示意图
最后,一条蛋白质被表示为一个 维的向量。如下式所示:
(4)
其中:
(5)
其中: 表示蛋白质序列中第i种氨基酸出现的概率, 表示蛋白质的第j层顺序因子, 表示顺序因子的权重因子。本文中取 ,最终一条膜蛋白序列被转换为一个50维的向量。
2.2.3. 拆分蛋白质二级结构组成(SSSC)
上述的两种特征提取方法都是从蛋白质序列中提取特征。然而,蛋白质的结构同样是重要的信息,蛋白质的功能取决于其局部结构性质,如3态和8态二级结构(SS3/SS8) [4]。对于3态二级结构,我们按照Cuff等人 [16] 定义的符号标准,E、H和C分别代表β-折叠、α-螺旋和其它结构。
在本文中,先使用Wang等人 [17] 开发的Raptorx服务器来获得原始蛋白质数据的3态二级结构数据。得到的结果为由E、H和C三个字母组成的序列。如表2所示,每个蛋白序列都转化为E、H和C三个字母组成的序列。
Table 2. Examples of 3-state secondary structures of proteins
表2. 蛋白质3态二级结构示例
获得蛋白质二级结构数据之后,需要对其进行特征提取。考虑到蛋白质的功能取决于其局部结构性质,结合SAAC的思想,本文将二级结构序列平均分成m个子序列,之后对这些子序列分别提取特征后再拼接融合成一个特征向量,以此来提取蛋白质的局部二级结构特征。对于其中的每个子序列,本文借鉴了二肽组成,四肽组成等方法,使用序列中n联体出现的频率来表示每个序列。经过上述的转换,一条蛋白质二级结构序列就被转换成 维的特征向量。
本文使用网格搜索方法来确定参数m和n的值,搜索范围为 ,,,并选取在随机森林默认参数下训练集十倍交叉验证结果最好的参数作为本文特征提取模型的参数。最终得到m和n的取值分别为3和5。
至此,本文将蛋白质3态二级结构序列中所有三联体出现的频率组成的向量称为蛋白质二级结构组成(SSC)。具体过程如公式(6)所示。
(6)
其中:L为蛋白质序列的长度, 定义为蛋白质3态二级结构序列中,三联体CCC出现的频率。可见,SSC将一条蛋白质转换为27维的特征向量。
最后将每条蛋白质二级结构序列平均拆分为5段子序列,并对每段子序列计算SSC,最后拼接为一个135维的特征向量。并将得到的特征向量称为拆分二级结构组成(SSSC)。
2.2.4. 特征融合
与PseAAC和SAAC特征只包含蛋白质序列信息不同,SSSC特征包含了蛋白质的二级结构信息。Zhang等人 [18] 的实验结果表明融合特征模型的分类效果优于原始特征模型。因此,在本节中,尝试将SSSC特征分别与PseAAC和SAAC特征进行拼接融合,得到两个新的特征来改进原PseAAC和SAAC特征以提高膜蛋白分类模型的分类效果。并将融合了SSSC和PseAAC特征的模型称为PseAAC-SSSC模型,类似的,将融合了SSSC和SAAC特征的模型称为SAAC-SSSC模型。PseAAC-SSSC模型和SAAC-SSSC模型得到的特征向量分别为195维和185维。
2.3. 分类算法
为了验证融合特征模型的有效性,本文使用随机森林、支持向量机、逻辑回归(LR)、多项朴素贝叶斯(MNB)、投票特征区间(VFI)、决策树(DT)以及KNN七种经典机器学习分类算法,进行了特征提取融合前后的对比实验。原因是机器学习方法速度快,对输入数据的维数没有要求,可以很好的验证不同特征的性能。最后基于Voting集成学习框架构建集成膜蛋白分类模型。
2.4. 评价指标
为了确保模型的可靠性,需要使用合适的方法对模型进行检验。本文采用十倍交叉验证来检验模型的可靠性,在十倍交叉验证中,将训练集数据随机平均分为十份,之后逐一选取一份作为测试样本,其余的作为训练样本,一共训练十次模型,最后选取十次实验结果的平均值作为模型评估指标。
本文使用的评价指标有:总体准确率(ACC)、类型准确率、马修斯相关系数(MCC)。
其中类型准确率的定义为某一种膜蛋白预测正确的数量与该类膜蛋白总数的比值。总体准确率定义如下:
(7)
其中: 和 分别表示第i种类型膜蛋白预测正确的个数和总个数。
马修斯相关系数计算公式如下:
(8)
其中:X和Y是两个矩阵,X表示每个蛋白质的预测类型,Y表示蛋白质的实际类型。 和 分别表示矩阵X和Y第j列的平均值。
3. 实验与结果分析
本文提出了一种新的膜蛋白特征提取方法,该方法在原有的蛋白质序列特征的基础上,加入了蛋白质二级结构信。为了验证新的特征方法的有效性,对比了新的蛋白质特征与原始的蛋白质序列特征在7种经典的机器学习算法下的分类效果。最后使用Voting集成学习框架,结合三个最优的分类算法并融合三个特征构建了最终的膜蛋白集成分类模型。
3.1. 参数选择
本文实验中训练的所有模型,都使用训练集数据训练模型参数,以十倍交叉验证的结果作为指标结合随机搜索或网格搜索对模型进行参数调优。
3.2. 特征提取方法的对比实验
基于PseAAC和SAAC两种原始序列特征,构建了两种融合特征模型PseAAC-SSSC和SAAC-SSSC。为了比较融合特征的性能,将这两种原始序列特征与两种融合特征分别与2.3节中的7种机器学习算法结合训练了28个膜蛋白分类模型,之后使用测试集数据的预测结果来评价模型的性能。表3列出28个膜蛋白分类模型的性能。
Table 3. The ACCs and MCCs of 28 models (%)
表3. 28个模型的ACC和MCC (%)
从表3可以看出,在所有特征模型下,基于随机森林和支持向量机的模型都获得了最高的ACC和MCC。这是因为随机森林是一种集成分类算法,由一系列决策树构成,在训练其中的决策树时,每次都随机抽取部分训练集样本以及部分特征进行训练,最后使用所有决策树的投票作为最终输出结果,随机森林解决了单个决策树易过拟合的问题,因此,随机森林获得了较好的分类效果。此外,膜蛋白分类问题本质上是一个非线性分类问题,膜蛋白序列中不同位置的氨基酸残基之间有一定的关联,提取到的特征向量中的不同值也会有关联,支持向量机的一个特性是能够挖掘非线性特征之间的相互作用,因此也获得了较好的结果。
Table 4. Type accuracy of PseAAC and PseAAC-SSSC under MNB (%)
表4. PseAAC和PseAAC-SSSC在多项式朴素贝叶斯下的类型准确率(%)
两种融合特征与原始序列特征的实验结果对比如图2和图3所示,可以看出,所有的基于融合特征的模型的ACC都高于基于单独序列特征的模型。几乎所有的融合特征模型的MCC高于对应的序列特征模型,只有PseAAC-SSSC模型在多项式朴素贝叶斯算法下的模型的MCC比PseAAC模型的MCC低0.21,通过观察这两个模型在8种类型上的准确率(见表4)发现,所有8种类型准确率中,融合特征模型获得了5个更高的类型准确率和2个更低的类型准确率。考虑到多项式朴素贝叶斯假设样本各属性相互独立,对输入特征的形式很敏感。在加入了蛋白质二级结构特征后,就可能存在部分的特征冗余,造成各类型准确率的波动,最终可能导致MCC发生变化。
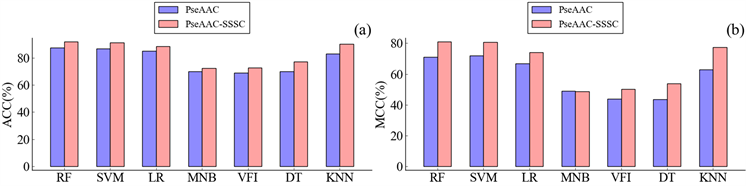
Figure 2. Contrasting experimental results between PseAAC and PseAAC-SSSC models
图2. PseAAC与PseAAC-SSSC模型的实验结果对比图

Figure 3. Contrasting experimental results between SAAC and SAAC-SSSC models
图3. SAAC与SAAC-SSSC模型的实验结果对比图
综上所述,融合特征模型相比于原始序列特征在膜蛋白分类效果上有较大的提升,从蛋白质二级结构中提取的特征可以作为蛋白质序列特征的一种补充。
3.3. 基于Voting集成学习的膜蛋白分类模型
在上一节中,比较了两种融合特征模型和对应的原始序列模型在7种不同分类算法下的性能,融合特征分类效果提升明显。因此,在本节中,尝试融合SAAC、PseAAC和SSSC三个特征。在所有特征模型中,随机森林、支持向量机、K近邻和逻辑回归四个模型的ACC和MCC最高。为了进一步提高模型的分类精度,集成学习方法是一个很好的思路。本文基于Voting集成学习框架构建膜蛋白集成学习分类模型。
Voting集成学习使用基分类器的投票作为最终结果,根据投票的方式可分为3类:绝对多数投票法、相对多数投票法和加权投票法。本文构建的Voting集成学习模型采用的是加权投票法,因为它使用基分类器输出的类概率进行投票,适用于基分类器较少的情况。
对于基分类器的选择,由于随机森林、支持向量机、K近邻和逻辑回归四个分类算法在上一节的融合特征实验中获得了前4的分类效果(表3所示),且与后3名分类器的实验结果拉开较大差距。因此,本文集成学习模型的基分类器从这4个里面选择。我们尝试了多种基分类器组合(包括4个基分类器的组合和随机森林、SVM和KNN 3个基分类器的组合以及它们的两两组合)。根据对比实验结果,最终选择随机森林、支持向量机和K近邻三个分类算法作为基分类器。至此,本文的Voting集成学习分类模型的框架如图4所示。
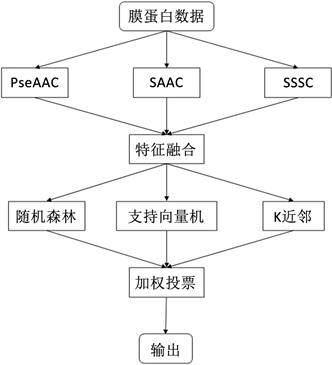
Figure 4. Flowchart of the ensemble learning classification model of this paper
图4. 本文的集成学习分类模型的流程图
训练集成学习模型时,先通过3.1中描述的参数选择方法单独训练每个基分类器的参数,之后训练三个基分类器的权重参数,这里由于集成模型训练速度缓慢,我们先基于随机森林和支持向量机构建Voting集成学习模型,并通过网格搜索法结合十倍交叉验证法训练随机森林和支持向量机两个基分类器的权重比值 ,搜索范围为 ,,最终得到当 时,模型的效果最好。之后在训练集成三个基分类器的模型的权重参数时,我们将随机森林和支持向量机的权重比例固定为 ,然后通过网格搜索法结合十倍交叉验证法在训练集上训练随机森林、支持向量机以及K近邻三个基分类器的权重比值 ,搜索范围为 , 且 ,最终得到当 时,十倍交叉验证的结果最优。最后,将测试集数据输入训练好的分类器中对模型进行评价。三个基分类器以及最终集成模型的ACC和MCC如表5所示。
从表5实验结果可以看出,对于三个基分类器,融合三种特征的特征融合模型相较于表3中融合两种特征的特征模型(PseAAC-SSSC和SAAC-SSSC模型)的ACC和MCC都有所提升。这说明了融合三种特征方案的有效性。Voting集成学习模型的ACC和MCC分别为94.02%和86.17%。
为了验证我们的集成模型在实际应用中的性能,我们和5种最先进的基于机器学习算法的膜蛋白分类模型作对比,所有在相同数据集上的测试集ACC如表6所示,我们的模型获得了最高的ACC。说明了本文模型的有效性。
Table 5. The ACCs and MCCs of 3 base classifiers and ensemble classifiers (%)
表5. 3个基分类器与集成分类器的ACC和MCC (%)
Table 6. Comparison to existing machine learning methods (%)
表6. 与现有的机器学习方法比较(%)
4. 总结
本文首次将蛋白质二级结构信息用于膜蛋白分类,首先提出了一种基于蛋白质二级结构信息的蛋白质特征提取方法,并将其融入两种现有的基于蛋白质序列的特征,得到两种融合特征。之后基于7种经典的机器学习分类算法,进行了一系列的膜蛋白分类对比实验。根据对比实验可以得出:基于融合特征与基于原始序列特征的机器学习模型分类比较,融合特征模型获得了更高的准确率,说明二级结构特征包含了许多序列特征没有的信息,与序列特征具有一定的互补性。因此,将蛋白质二级结构特征用于膜蛋白分类可以有效提高预测的精度,具有一定的研究价值。最后,融合三种特征,基于Voting集成学习框架,集成三种机器学习算法,建立了一个膜蛋白分类模型。并与现有的几种基于机器学习的膜蛋白分类模型进行比较,结果表明,本文的模型优于其它机器学习模型。
本文使用的是机器学习的方法,随着技术的进步,神经网络已被广泛用于解决实际问题。因此,在后续的研究中,可以在集成学习框架中加入神经网络的方法。
文章引用
苏鹏程. 基于融合特征和Voting集成学习的膜蛋白类型预测
Prediction of Membrane Protein Types Based on Fusion Feature Information and Voting Ensemble Learning[J]. 计算生物学, 2021, 11(04): 49-58. https://doi.org/10.12677/HJCB.2021.114006
参考文献
- 1. Almén, M.S., Nordström, K.J., Fredriksson, R. and Schiöth, H.B. (2009) Mapping the Human Membrane Proteome: A Majority of the Human Membrane Proteins Can Be Classified According to Function and Evolutionary Origin. BMC Bi-ology, 7, Article No. 50. https://doi.org/10.1186/1741-7007-7-50
- 2. Overington, J.P., Al-Lazikani, B. and Hop-kins, A.L. (2006) How Many Drug Targets Are There? Nature Reviews Drug Discovery, 5, 993-996. https://doi.org/10.1038/nrd2199
- 3. Chou, K.C. and Shen, H.B. (2007) MemType-2L: A Web Server for Predict-ing Membrane Proteins and Their Types by Incorporating Evolution Information through Pse-PSSM. Biochemical and Biophysical Research Communications, 360, 339-345. https://doi.org/10.1016/j.bbrc.2007.06.027
- 4. Chou, K.C. and Elrod, D.W. (1999) Prediction of Membrane Protein Types and Subcellular Locations. Proteins: Structure Function and Bioinformatics, 34, 137-153. https://doi.org/10.1002/(SICI)1097-0134(19990101)34:1<137::AID-PROT11>3.0.CO;2-O
- 5. Chou, K.C. (2001) Prediction of Protein Cellular Attributes Using Pseudo-Amino Acid Composition. Proteins: Structure Function and Bio-informatics, 43, 246-255. https://doi.org/10.1002/prot.1035
- 6. Hayat, M., Khan, A. and Yeasin, M. (2012) Pre-diction of Membrane Proteins Using Split Amino Acid and Ensemble Classification. Amino Acids, 42, 2447-2460. https://doi.org/10.1007/s00726-011-1053-5
- 7. Petrilli, P. (1993) Classification of Protein Sequences by Their Dipeptide Composition. Bioinformatics, 9, 205-209. https://doi.org/10.1093/bioinformatics/9.2.205
- 8. Alphonse, A.S., Mary, N.A.B. and Starvin, M.S. (2020) Clas-sification of Membrane Protein Using Tetra Peptide Pattern. Analytical Biochemistry, 606, Article ID: 113845. https://doi.org/10.1016/j.ab.2020.113845
- 9. Hayat, M. and Khan, A. (2012) Mem-PHybrid: Hybrid Fea-tures-Based Prediction System for Classifying Membrane Protein Types. Analytical Biochemistry, 424, 35-44. https://doi.org/10.1016/j.ab.2012.02.007
- 10. Wang, H., Ding, Y.J., Tang, J.J. and Guo, F. (2020) Identification of Membrane Protein Types via Multivariate Information Fusion with Hilbert-Schmidt Independence Criterion. Neurocom-puting, 83, 257-269. https://doi.org/10.1016/j.neucom.2019.11.103
- 11. Wang, L.P., Yuan, Z.T., Chen, X.H. and Zhou, Z.F. (2010) The Prediction of Membrane Protein Types with NPE. IEICE Electronics Express, 7, 397-402. https://doi.org/10.1587/elex.7.397
- 12. Hayat, M. and Khan, A. (2010) Predicting Membrane Protein Types by Fusing Composite Protein Sequence Features into Pseudo Amino Acid Composition. Journal of Theoretical Biology, 271, 10-17. https://doi.org/10.1016/j.jtbi.2010.11.017
- 13. 郭磊, 王顺芳. 序列信息融合与两阶段特征选择的膜蛋白预测[J]. 计算机工程与应用, 2019, 55(6): 145-150.
- 14. Myers, J.K. and Oas, T.G. (2001) Preorganized Secondary Structure as an Important Determinant of Fast Protein Folding. Nature Structural Biology, 8, 552-558. https://doi.org/10.1038/88626
- 15. Wan, S.B., Mak, M.-W. and Kung, S.-Y. (2016) Benchmark Data for Identify-ing Multifunctional Types of Membrane Proteins. Data in Brief, 8, 105-107. https://doi.org/10.1016/j.dib.2016.05.024
- 16. Cuff, J.A. and Barton, G.J. (1999) Evaluation and Improvement of Multiple Sequence Methods for Protein Secondary Structure Prediction. Proteins: Structure Function and Bioinformatics, 34, 508-519. https://doi.org/10.1002/(SICI)1097-0134(19990301)34:4<508::AID-PROT10>3.0.CO;2-4
- 17. Wang, S., Li, W., Liu, S.W. and Xu, J. (2014) RaptorX-Property: A Web Server for Protein Structure Property Prediction. Nucleic Acids Research, 44, W430-W435. https://doi.org/10.1093/nar/gkw306
- 18. Zhang, X.L. and Chen, L. (2020) Prediction of Membrane Protein Types by Fusing Protein-Protein Interaction and Protein Sequence Information. BBA-Proteins and Proteomics, 1868, Article ID: 140524. https://doi.org/10.1016/j.bbapap.2020.140524
- 19. Huang, G.H., Zhang, Y.C., Chen, L., Zhang, N., Huang, T. and Cai, Y.-D. (2014) Prediction of Multi-Type Membrane Proteins in Human by an Integrated Approach. PLOS ONE, 9, e93553. https://doi.org/10.1371/journal.pone.0093553
- 20. Nanni, L., Brahnam, S. and Lumini, A. (2012) Wavelet Images and Chou’s Pseudo Amino Acid Composition for Protein Classification. Amino Acids, 43, 657-665. https://doi.org/10.1007/s00726-011-1114-9
- 21. Chen, Y.K. and Li, K.B. (2013) Predicting Membrane Protein Types by Incorporating Protein Topology, Domains, Signal Peptides, and Physicochemical Properties into the General form of Chou’s Pseudo Amino Acid Composition. Journal of Theoretical Biology, 318, 1-12. https://doi.org/10.1016/j.jtbi.2012.10.033