Advances in Condensed Matter Physics
Vol.3 No.04(2014), Article
ID:14373,6
pages
DOI:10.12677/CMP.2014.34006
Effects of p-Orbital On-Site Coulumb Repulsion on the Electronic Properties of Mg2Sn
1Department of Physics, Tianjin Polytechnic University, Tianjin
2School of Physics, Peking University, Beijing
Email: *liuguanghua@tjpu.edu.cn
Copyright © 2014 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received: Sep. 22nd, 2014; revised: Oct. 24th, 2014; accepted: Oct. 30th, 2014
ABSTRACT
The band structure, density of states, electron density difference and dielectric function of Mg2Sn are investigated systematically by using the generalized gradient approximation based on density functional theory. To describe the strong electron correlation in the Sn 5p states, the GGA plus on-site repulsion method (GGA+U) is used in the electrical properties calculation of Mg2Sn. It is found that the band gap is negative with the effective U value of 0 eV, which is quite different from the experimental result. As the effective U value of 1.88 eV is considered, the compound is a semiconductor with an indirect band gap of 0.30 eV, which agrees with the previous experimental observation satisfactorily. The calculations of electron density difference show that the covalent bond between Sn-Sn is stronger than the covalent bond between Sn-Mg, which is accordant with the practical situation. The calculation of dielectric function agrees well with the experimental result, too. The important influence of the p-orbital on-site Coulumb repulsion for the electronic properties of Mg2Sn is confirmed.
Keywords:GGA + U, On-Site Coulumb Repulsion, Electronic Properties

p轨道占位库仑排斥对Mg2Sn电学性质的影响
张 钰1,孔龙娟1,强 凌2,刘光华1*
1天津工业大学理学院,天津
2北京大学物理学院,北京
Email: *liuguanghua@tjpu.edu.cn
收稿日期:2014年9月22日;修回日期:2014年10月24日;录用日期:2014年10月30日

摘 要
利用基于密度泛函理论的广义梯度近似方法计算了Mg2Sn的能带结构、能态密度、差分电荷密度和介电函数。为了考虑Sn中5p轨道的强电子关联效应,我们采用GGA + U的方法计算Mg2Sn的电学性质。当有效U值为0 eV时,Mg2Sn带隙为负,与实验值相差较大。当有效U值为1.88eV时,间接带隙为0.30 eV,与实验值吻合较好。差分电荷密度的计算表明Mg2Sn中Sn-Sn键相对于Sn-Mg键有着较强的共价性,符合实际情况,介电函数的计算结果也与实验结果基本一致。这些证实p轨道占位库仑排斥对Mg2Sn的电学性质有着较大的影响。
关键词
GGA + U,占位库仑排斥,电学性质

1. 引言
具有反萤石结构的半导体Mg2X (X = Si, Ge, Sn)热电组件因为具有塞贝克效应、无毒、廉价、低电阻率、低热导率、低密度等特点[1] [2] ,在废热回收及太阳热能设备等领域有着广泛的应用[3] [4] ,因此近几十年来在理论与实验中对它们的探索一直没有中断。J. BOUREOIS等研究了在不同温度下Mg2X的结构、电子系数及声子谱等特性的理论与实验结果[1] ,Fei Yu等研究了Mg2X在不同压强下其结构发生的改变及相应的能态密度与光学性能的变化[5] ,K. Kutorasinski则使用KKB算法发现Mg2X在价带Γ上的自旋轨道相互作用大部分取决于X原子[6] 。A. Stella等则通过实验测定了Mg2X的带隙宽度[7] 。但是,通过大量的文献我们发现在相关的理论研究中Mg2Si与Mg2Ge的带隙与实验值较为吻合[1] [7] [8] ,但在有关Mg2Sn的带隙的理论计算却与实验值[7] [9] -[11] 有较大的差距。
在第一性原理的计算中,对存在库仑排斥效应的半导体,模拟计算时仅使用LDA或GGA算法不能准确预测其性质,其中带隙的理论计算值与实验值有着较大的差异[12] 。基于密度泛函理论与局域自旋密度近似的计算不能准确得到预期结果,其主要原因有两点:一是计算中过多的自旋极化空穴移位导致材料体现出半金属基态和室温下的铁磁性性质;二是无法准确计算出材料在基态的性质[13] 。在密度泛函理论的计算中Kohn-Sham方程的平均场特点和对强库仑相关作用的忽视更会导致计算结果与预期值的偏离,但文献[14] 指出,在计算中加入Hubbard参数U则能够很好的修正这些问题。
由于人们通常认为占位库仑排斥主要是3d和4f轨道的特性,因此p轨道的占位关联效应容易被忽略[15] 。但是最近有关d0磁性材料的研究表明,材料的磁性来自半满的p轨道[13] [16] 。文献[17] [18] 用GGA + U的方法计算表明RbO2的d轨道没有电子,其磁性主要来自于氧元素p轨道的贡献。除此之外,用GGA + U对d10类型的ZnO电学性质的研究表明,当考虑了O元素2p轨道的占位库仑排斥后理论值与实验值基本一致[19] ,表明p轨道的占位库仑排斥效应非常重要。受到以上研究的启发,我们猜想p轨道的占位库仑排斥对Mg2Sn的电学性质也可能有较大影响。为此本文将把p轨道的占位库仑排斥效应考虑进来研究Mg2Sn的电学性质。
2. 计算方法
具有反萤石结构的Mg2Sn空间群为Fm3m,晶格常数a = 6.81Å,Sn原子占据点(0,0,0),Mg原子占据点(1/4, 1/4, 1/4),晶胞模型[20] 如图1所示。
文中所有计算工作都是在Material Studio软件中的CASTEP (Cambridge serial total energy pack-age)软件包中完成[21] 。它是建立在第一性原理的基础上,采用密度泛函理论(DFT)和分子动力学理论对材料进行分析的软件包。在计算中选择广义梯度近似(GGA)下的Perdew-Wang (PW91)来处理电子间的交换关联能,对于价电子与原子实之间的相互作用选择超软赝势在倒格子空间中进行计算。先对Mg2Sn晶胞模型进行几何优化,得到最优结构后再进行能带结构的计算。调节U的值根据实验值[7] 找到最佳的Hubbard参数U后再计算相关的结构参数,电学性质与光学特性。设置能带截断值为330 eV,自洽计算(SCF)的收敛精度为2.0 × 10−6 eV/atom,布里渊区的K点取为4 × 4 × 4,K点间隔为0.5 nm−1。
3. 结果与讨论
3.1. Hubbard参数U
在理论计算中,对Mg2Sn使用广义梯度近似(GGA)下的Perdew-Wang (PW91)函数时计算的带隙非常微小[22] ,使用fully relativistic (FR)算法与semirelativistic (SR) KKR算法时带隙值分别为−0.25 eV与−0.17 eV[6] ,均与实验值差距较大。我们猜想这是由于忽略了p轨道强电子关联所导致的结果,所以考虑到p轨道的占位库仑排斥效应后我们采用GGA + U的方法计算Mg2Sn的电学性质。计算表明Mg2Sn的带隙随着有效U值得增加而增加,如图2所示。当Hubbard参数U值为1.88 eV,Mg2Sn的带隙为0.30 eV,与实验值[7] 吻合得较好。由此我们可以确定有效U值为1.88 eV。
3.2. 能带结构和态密度分析
为了研究Mg2Sn晶体中Sn原子p轨道上的占位库仑排斥效应对结构中原子间相互作用的影响,计算了Mg2Sn在不引入占位库仑排斥效应与引入占位库仑排斥效应下的能带结构、总体态密度和局域态密度。能带结构如图3所示,其中虚线表示费米能级。由图可以看出Mg2Sn能带价带的最高点和导带的最低点不在同一个K点处,说明Mg2Sn的带隙为间接能隙。在不考虑p轨道的占位库仑排斥效应的情况下,计算得出Mg2Sn带隙为负,如图3(a)所示;引入占位库仑排斥效应并且Hubbard参数U取1.88 eV时,Mg2Sn带隙为0.30 eV如图3(b)所示,与实验值吻合得较好。
图4为Mg2Sn在不考虑占位库仑排斥效应时的总态密度和Mg,Sn各亚层电子的能态密度。可以看出Mg2Sn在费米能级处有一定的态密度值,说明它拥有金属的特性[23] 。根据对总态密度和各原子分态密度的分析,Mg2Sn在低于费米能级的能量区域内,其总态密度主要来源于Sn-5s态和Sn-5p态,在费米能级以上的能量区域,Mg2Sn的总态密度主要贡献来自Sn-5s态,Sn-5p态和Mg-2p态。
在考虑占位库仑排斥效应时Mg2Sn的总态密度和Mg,Sn各亚层电子的能态密度时,结果如图5所示,可以发现,在费米能级处Mg2Sn有一个赝能隙[24] ,说明Mg2Sn中有共价键。同样,在低能量价带范围,Mg2Sn总态密度主要来源于Sn-5s态和Sn-5p态,在费米能级以上的能量区域,Mg2Sn的总态密度的主要贡献来自Mg-2p态和Sn-5p态。因此对于Mg2Sn其Sn-5p态对总态密度的贡献占主导作用。
3.3. 差分电荷密度
为了更清晰的探明Mg2Sn的成键特征,我们研究了它的差分电荷密度,如图6所示,可以看出Mg2Sn中Sn原子之间有大量的电荷发生交叠,说明Sn-Sn键是强烈的共价键,但Sn-Mg原子间的粘合强度相对较弱,说明Sn-Mg键是较弱的共价键。这与其他研究对Mg2Sn电荷密度的分析的结论[22] 相同,说明
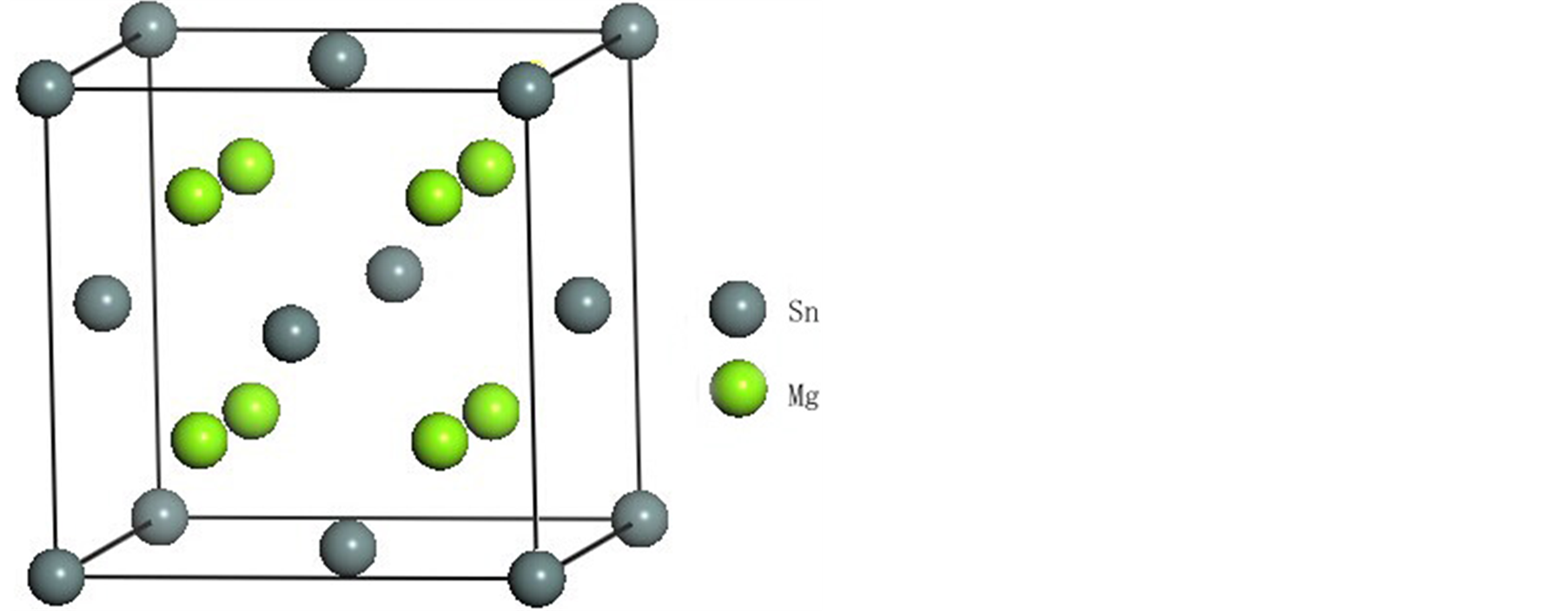
Figure 1. Crystal structure of Mg2Sn
图1. Mg2Sn的晶胞结构

Figure 2. The effective U value dependence of the band gaps of Mg2Sn
图2. 加入Hubbard参数U后Mg2Sn带隙的变化

Figure 3. The energy bands without and with the Hubbard U: (a) Mg2Sn (U = 0 eV);(b) Mg2Sn (U = 1.88 eV)
图3. 加入Hubbard参数U前后Mg2Sn的能带结构:(a) Mg2Sn (U = 0 eV);(b) Mg2Sn (U = 1.88 eV)

Figure 4. The DOS and PDOS of Mg2Sn with U = 0 eV
图4. 当Hubbard参数U = 0 eV时Mg2Sn的DOS与PDOS
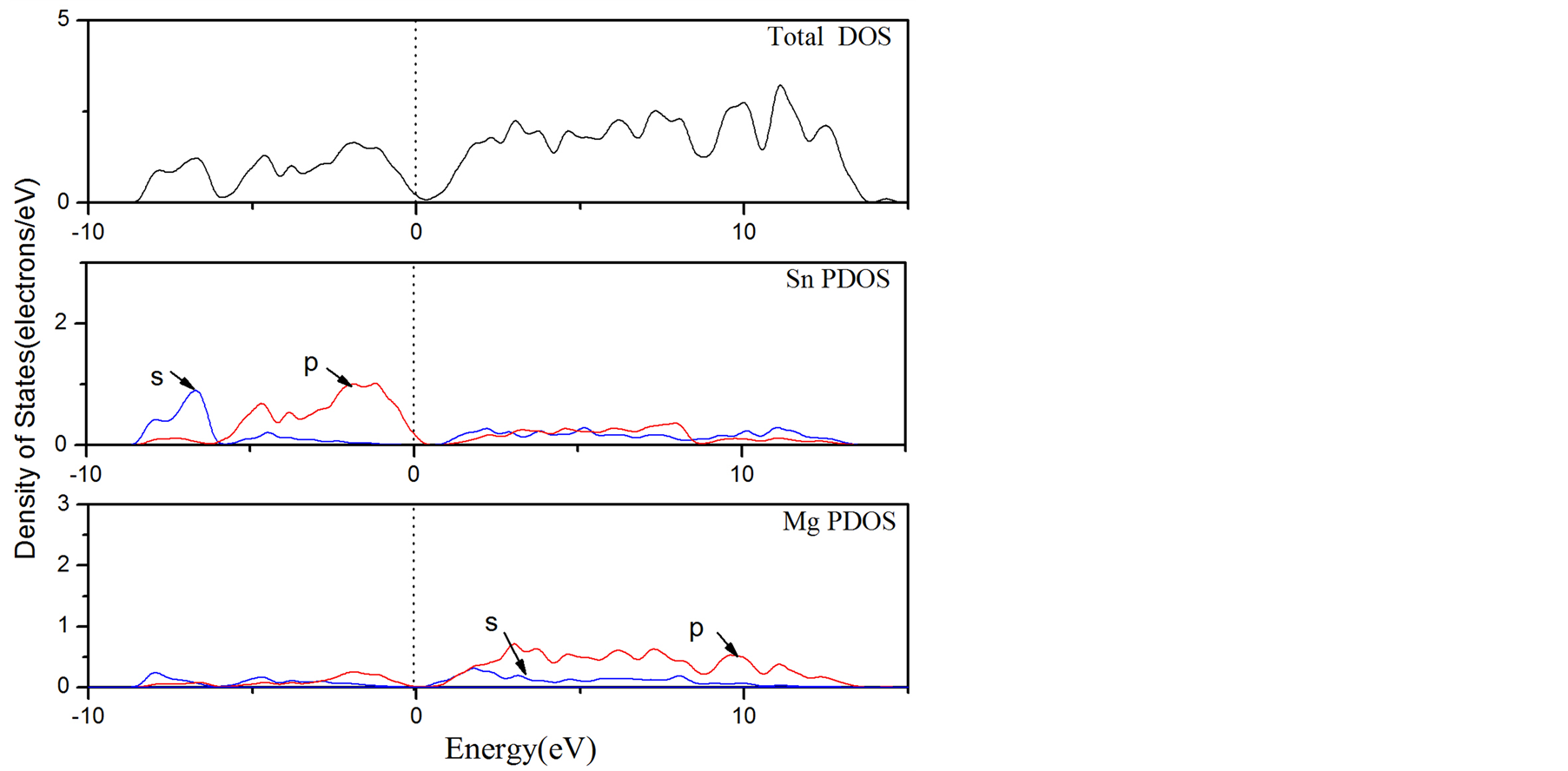
Figure 5. The DOS and PDOS of Mg2Sn with U = 1.88 eV
图5. 当Hubbard参数U = 1.88 eV时Mg2Sn的DOS与PDOS
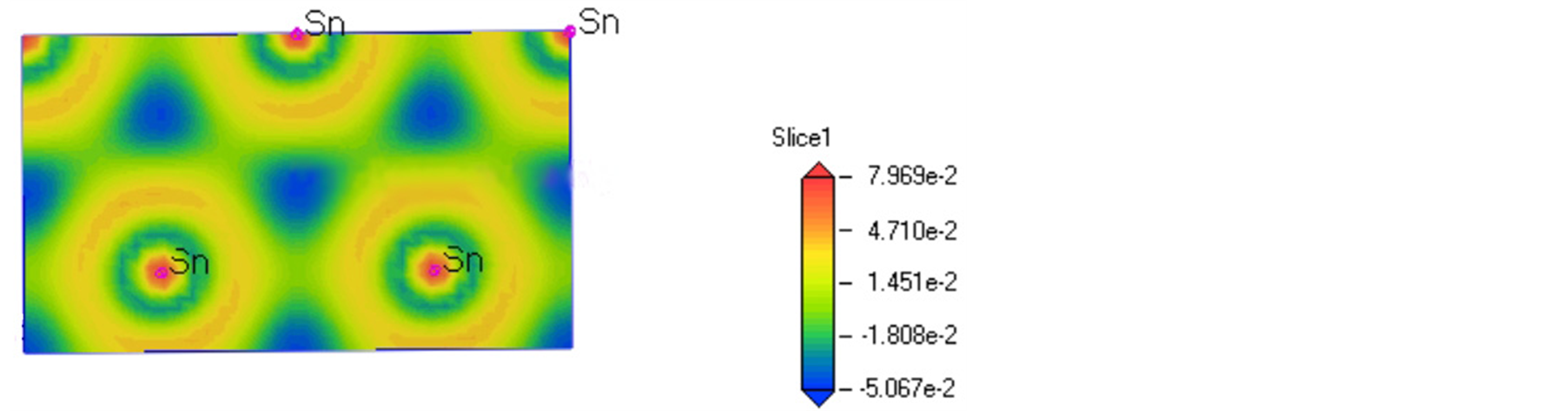
Figure 6. Electron density difference in the plane (110) of Mg2Sn
图6. Mg2Sn在(110)面上的差分电荷密度分布

Figure 7. Theoreticaland experimental dielectric function imaginary part 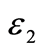 of Mg2Sn
of Mg2Sn
图7. Mg2Sn介电函数虚部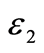 的理论计算结果和实验结果
的理论计算结果和实验结果
了Sn-Sn共价键对Mg2Sn有着强烈影响,Mg2Sn有着较强的结构稳定性。
3.4. 介电函数
介电函数能够反映固体电子结构与带间跃迁微观物理过程的关系,它能够反映出固体能带结构及其他各种光谱信息[25] 。在线性响应范围内,研究物体的光学性能时经常使用光的复介电函数方程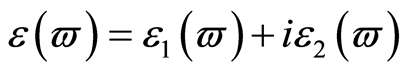 或复折射率
或复折射率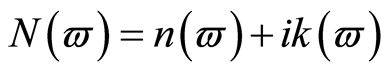 来描述。其中
来描述。其中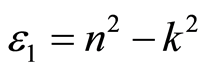 ,
,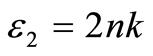 ,n和k分别为反射系数和消光系数。
,n和k分别为反射系数和消光系数。
根据直接跃迁几率的定义和克拉默斯-克勒尼希色散关系我们可以直接推导出晶体介电函数虚部、实部、吸收系数、反射系数等[25] [26] 。图7为理论计算的Mg2Sn的介电函数虚部![]() 与实验值[27] 的曲线图。可以看出实验值与计算值吻合度较高,表明利用剪刀算符进行修正是合理的。介电函数虚部
与实验值[27] 的曲线图。可以看出实验值与计算值吻合度较高,表明利用剪刀算符进行修正是合理的。介电函数虚部![]() 在能量为2.5 eV、5.6 eV处出现介电峰,且主要的跃迁峰都发生在大于2.0 eV的能量区,说明Mg2Sn的介电峰主要来自间接跃迁。
在能量为2.5 eV、5.6 eV处出现介电峰,且主要的跃迁峰都发生在大于2.0 eV的能量区,说明Mg2Sn的介电峰主要来自间接跃迁。
4. 结论
利用基于密度泛函理论的广义梯度近似方法研究了Mg2Sn在引入占位库仑排斥效应后的能带结构,态密度,差分电荷密度和介电函数。计算结果表明Mg2Sn是一种间接带隙半导体材料,其中Sn原子p轨道对Mg2Sn有着较强的影响,因此选择对Mg2Sn中Sn原子p轨道加入Hubbard参数U,使用GGA + U方法进行计算,根据计算结果确定Hubbard参数U = 1.88 eV。在加入Hubbard参数U后对Mg2Sn的态密度,差分电荷密度和介电函数进行计算,发现其与其他计算结果和实验值几乎完全一致,证明了p轨道占位库仑排斥效应对Mg2Sn的电学性质有着不可忽视的影响。
基金项目
感谢国家自然基金项目(11347008)的资助。
参考文献 (References)
- [1] Bougreios, J., Tobola, J., Wiendlocha, B., Chaput, L., Zwolenski, P., Recour, Q. and Scherrer, H. (2013) Study of election, phonon and crystal stability versus thermoelectric properties in Mg2X(X = Si, Sn) compounds and their alloys. Functional Materials Letters, 6, 1340005.
- [2] Noda, Y., Kon, H., Furukawa, Y., Otsuka, N., Nishida, I.A. and Masumoto, K. (1992) Temperature dependence of thermoelectric properties of Mg2Si0.6Ge0.4. Materials Transactions. JIM, 33, 851-855.
- [3] Snyder, J.G. and Toberer, E.S. (2008) Complex thermoelectric materials. Nature Materials, 7, 105-114.
- [4] Daniel, K., Bed, P., Hsien-Ping, F., Caylor, J.C., Yu, B., Yan, X., Ma, Y., Wang, X.W., Wang, S.Z., Muto, A., Kenneth, M., Matteo, C., Ren, Z.F. and Chen, G. (2011) High-performance flat-panel solar thermoelectric generators with high thermal concentration. Nature Materials, 10, 532-538.
- [5] Yu, F., Sun, J.X. and Chen, T.H. (2011) High-pressure phase transitions of Mg2Ge and Mg2Sn: First-principles calculations. Physica B, 406, 1789-1794.
- [6] Kutorasinski, K., Wiendlocha, B., Tobola, J. and Kaprzyk, S. (2014) Importance of relativistic effects in electronic structure and thermopower calculations for Mg2Si, Mg2Ge, Mg2Sn. Physical Review B, 89, 115205.
- [7] Stella, A., Brothers, A.D., Hopkins, R.H. and Lynch, D.W. (1967) Pressure coefficient of the band gap in Mg2Si, Mg2Ge, and Mg2Sn. physica Status Solidi, 23, 697-702.
- [8] Premlata, P. and Sankar, P.S. (2011) First principles study of electronic,elastic and lattice dynamical properties of Mg2X (X = Si, Ge and Sn) compounds. Indian Journal of Pure & Applied Physics, 49, 692-697.
- [9] Buchenauer, C.J. and Cardona, M. (1971) Raman Scattering in Mg2Si, Mg2Ge, and Mg2Sn. Physical Review B, 3, 2504-2507.
- [10] Scouler, W.J. (1969) Optical properties of Mg2Si, Mg2Ge and Mg2Sn from 0.6 to 11.0 eV at 77 degK. Physical Review, 178, 1353-1357.
- [11] Blunt, R.F., Frederikse, H.P.R. and Hosler, W.R. (1955) Electrical and optical properties of intermetallic compounds IV magnesium stannide. Physical Review, 100, 663-666.
- [12] Karazhanov, S.Z., Ravindran, P., Kjekshus, A., Fjellvag, H., Grossner, U. and Svensson, B.G. (2006) Coulomb correlation effects in zinc monochalcogenides. Journal of Applied Physics, 100, Article ID: 043709.
- [13] Droghetti, A., Pemmaraju, C.D. and Sanvito, S. (2008) Predicting d0 magnetism: Self-interaction correction scheme. Physical Review B, 78, Article ID: 140404(R).
- [14] Vladimir, I.A., Jan, Z. and Ole, K.A. (1991) Band theory and Mott insulators: Hubbard U instead of Stoner I. Physical Review B, 44, 943-954.
- [15] Ylvisaker, E.R., Singh, R.R.P. and Picktt, W.E. (2010) Orbital order, stacking defects, and spin fluctuations in the p-electron molecular solid RbO2. Physical Review B, 81, Article ID: 180405(R).
- [16] Coey, J.M.D. (2005) d0 ferromagnetism. Solid State Sciences, 7, 660-667.
- [17] Kovacik, R. and Ederer, C. (2009) Correlation effects in p-electron magnets: Electronic structure of RbO2 from first principles. Physical Review B, 80, Article ID: 140411.
- [18] Mim, K., Kim, B.H., Choi, C.H. and Min, B.I. (2009) Antiferromagnetic and structural transitions in the superoxide KO2 from first principles: A 2p-electron system with spin-orbital-lattice coupling. Physical Review B, 81, Article ID: 100409(R).
- [19] Deng, X.Y., Liu, G.H., Jing, X.P. and Tian, G.S. (2014) On-site correlation of p-electron in d10 semiconductor zinc oxide. International Journal Quantum Chemistry, 114, 468-472.
- [20] Duman, S., Tutuncu, H.M., Bagci, S. and Srivastava, G.P. (2007) Ab initio determination of structural and dynamical properties of Mg2Sn. AIP Conference Proceedings, 899, 247-248.
- [21] Segall, M.D., Lindan, P.L.D., Probert, M.J., Pickard, C., Hasnip, P.J., Clark, S.J. and Payne, M.C. (2002) First-principles simulation: Ideas, illustrations and the CASTEP code. Journal of Physics-Condensed Matter, 14, 2717- 2744.
- [22] 李鹏博, 潘荣凯, 马丽, 王明辉, 卞楠, 唐壁玉, 彭立明, 丁文江 (2014) Mg2Sn的弹性性能和电子结构的第一性原理计算. 广西大学学报(自然科学版), 3, 479-483.
- [23] Wang, J.Y. and Zhou, Y.C. (2004) Polymorphism of Ti3SiC2 ceramic: First-principles investigations. Physical Review B, 69, Article ID: 144108.
- [24] Ravindran, P., Fast, L., Korzhavyi, P.A., Johansson, B., Wills, J. and Eriksson, O. (1998) Density functional theory for calculation of elastic properties of orthorhomcic crystals: Applications to TiSi. Journal of Applied Physics, 84, 4891- 4904.
- [25] 沈学础 (2002) 半导体光谱和光学性质. 科学出版社,北京.
- [26] 方容川 (2001) 固体光谱学. 中国科学技术大学出版社,合肥.

NOTES
*通讯作者。