Biophysics
Vol.
06
No.
04
(
2018
), Article ID:
26849
,
9
pages
10.12677/BIPHY.2018.6464007
A New Method for Protein Complex Detection Based on Multi-Source Information
Yang Yu
Software College, Shenyang Normal University, Shenyang Liaoning

Received: Aug. 31st, 2018; accepted: Sep. 11th, 2018; published: Sep. 18th, 2018

ABSTRACT
Protein complexes are important biological function modules in protein interaction (PPI) network and play an important role in biological activities. Usually the topological structure is used as the condition for clustering in the PPI network, which constrains the overall performance of complex identification. To improve this problem, a new complex identification algorithm is proposed based on fusion of multi-source information. Firstly, based on the semantic similarity of GO, we construct a weighted PPI model and design a core-attachment clustering algorithm based on the density, diameter and cosine similarity. Secondly, the seed nodes are sequentially selected according to the descending order of the node degrees in the network and expanded in weighted graph model to identify protein complex. Lastly, the proposed method is compared with three classical algorithms. The experimental results indicate that the algorithm can effectively enhance the performance in terms of precision, F-measure and functional enrichment analysis.
Keywords:Multi-Source Information, Weighted Graph Model, Sequence Information, Complex Identification
一种新的基于多源信息的复合物 识别算法
于杨
沈阳师范大学软件学院,辽宁 沈阳
收稿日期:2018年8月31日;录用日期:2018年9月11日;发布日期:2018年9月18日

摘 要
蛋白质复合物是生物网络中一个重要的生物功能模块,在生物活动中起着重要的作用。通常在PPI中仅利用拓扑结构作为复合物识别的条件,约束了复合物识别的综合性能。针对此问题,本文提出一种新的基于多源信息的复合物识别算法。首先,基于GO的语义相似性构建有权平均蛋白质网络模型,然后设计以稠密子图的密度、直径和余玄相似度为聚类条件的核–附属的聚类算法;其次,根据网络中节点度值降序依次选择种子节点,并且在有权网络模型中对种子节点进行扩展以识别蛋白质复合物。最后,本文将其与CFinder,MCode和MCL进行比较和分析,实验结果表明,本文提出的算法在准确率,F度量和功能富集分析的方面均能有效地提高复合物识别的性能。
关键词 :多源信息,有权图模型,序列信息,复合物识别

Copyright © 2018 by author and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 引言
在各类生物网络系统中,模块化是一个普遍存在的现象 [1] 。生物系统是一个模块化的组织,由不同的生物功能模块组成。大部分细胞的活动体现在网络的相互作用的模块中,改变不同的模块之间的连接可能会影响细胞特性和功能的变化。蛋白质复合物是指在时间和空间上通过相互作用的蛋白质集合。研究发现在蛋白模块内部细胞功能和生化活动相互协调实现特定的生物功能,因此识别蛋白质相互作用网络(PPI网络)中模块化结构对于理解和分析生物功能具有重要的医学意义 [2] 。
基于图论知识,PPI网络可以用图表示,图中的点对应于网络中的蛋白质,边对应于蛋白质之间的相互作用关系。蛋白质复合物识别的任务即是利用以聚类技术为基础的计算方法,挖掘网络中不同的子图(复合物)。PPI网络是一种高度动态和结构化的复杂网络,它具有无尺度分布、小世界性质和功能模块化的拓扑特征。无尺度分布指PPI网络中连接度为k的节点出现的概率满足幂律分布;小世界性质指PPI网络所含的蛋白质节点具有较短的平均最短路径长度和较高的平均聚集系数 [1] ;功能模块化则指在PPI网络中相互连接密集的蛋白质形成一个模块,并完成同样的功能。这些不同的结构特征制约或影响着功能模块检测过程和方法的设计,尤其网络模块化的特征更是许多检测方法实现的理论依据。
蛋白质复合物主要有三个特点:1) 具有较高的密度 [3] ;2) 具有核心–附属功能特征,核心功能由一些连接高度紧密的具有共同表达和功能相似性蛋白质组成,行使并完成复合物的主要功能,附属则是辅助核心功能完成特定的生物功能;3) 重叠性,即指一个蛋白质可以参与到多个复合物中执行不同的生物功能 [4] 。
传统方法通过生物实验检测蛋白质复合物,但该方法在检测费用、时间和质量上具有局限性。因此,以机器学习、数据挖掘和大数据智能分析为理论基础,识别PPI网络中模块的方法迅速兴起。新的思想和检测方法不断涌现,Nature、Science、Proceedings of National Academy of Sciences (PNAS)、Nucleic Acids Research、Bioinformatics等国际权威期刊以及许多国际会议都多次探讨相关研究,在生物信息领域掀起了利用计算方法在PPI网络中预测蛋白质复合物的研究热潮。根据所采用的计算模型和机理,蛋白质复合体预测方法主要有两大类。第一类方法主要基于PPI网络拓扑的预测方法,例如MCODE [5] ,CFinder [6] ,MCL [7] ,L [8] ,SEGC [9] 。MCODE检测网络中链接相对密集的簇为蛋白质复合物。Adamcsek等人基于团渗透方法设计在蛋白质相互作用网络中预测蛋白质复合物的CFiner工具。MCL基于expansion和inflation两个操作在有权或无权网络上识别蛋白质复合物。L方法通过基于边的层次聚类识别重叠的蛋白质复合物。SEGC基于种子扩展图聚类算法检测蛋白质复合物。生物实验技术产生的PPI数据通常具有较高的假阳性、假阴性和不完整性,这使得基于拓扑信息预测复合物在准确率和拓扑结构的多样性方面存在不足。第二类融合其他生物信息的方法,常见的多源生物数据包括:基因表达数据(时间、空间、特定环境等各种不同基因表达数据)、功能信息数据、免疫共沉淀数据、结构域等。事实上,相互作用的蛋白质可能具有相似的基因表达谱,此类方法如BiCAMWI [10] 、CPredictor 3.0 [11] 。BiCAMWI融合基因表达数据构建动态网络预测蛋白质复合物。CPredictor 3.0融合基因表达数据和蛋白质功能注释信息预测蛋白质复合物。已有的多源生物信息融合方法虽然考虑到生物信息来源的多样性,但是具体对每类生物信息分析得不够清晰,且较少考虑到生物信息本身的可靠性和真实性。本文提出一种融合多源生物信息构建有权模型识别复合物的新方法,具体研究内容包括:设计复合物多维特征表示方法;融合酵母蛋白质相互作用数据和基因本体信息,实现有权网络模型;提出基于核–附属结构的复合物预测算法;比较并分析MWCA与三个经典的方法,实验结果表明,MWCA在召回率、f度量和功能富集分析方面的表现均显著优于CFinder,MCode和MCL。
2. 基于多源信息的复合物识别算法(MWCA)
基于PPI网络的研究通常将PPI网络表示为无向图 ,其中V对应于PPI网络中蛋白质节点集合,E代表PPI网络中两个蛋白质之间存在相互作用关系集合。
2.1. 融合基因本体的有权网络模型
在研究中发现,同一物种的不同类型的生物数据之间存在良好的互补性 [12] 。本文融合基因本体信息构建有权网络模型。基因本体(GO)是用于解释生物实验结果的最常用的生物信息学资源之一。GO提供结构化的、可控的词汇表来描述基因的三种属性,即分子功能、生物过程和细胞组成。GO提供了一种方便而重要来研究函数相似度的方法。基于GO的语义相似度已经有成功地应用于许多研究领域,如基因功能预测,基因网络分析,同源性分析,基因关联可视化等。因而,酵母的基因本体数据能在相当大的程度上弥补PPI数据的不足。
本文利用GO术语计算DIP网络中两个蛋白质被注释的集合之间的功能相似性,统计被 GO 注释后的两个蛋白质对应的所有术语之间的最佳匹配策略,计算被GO注释后的两个蛋白质对应的所有术语时间的语义相似性的平均值作为图的权值,构建有权图模型,如公式(1)~(3),其中 和 分别表示DIP数据中蛋白质X和Y对应的GO术语集合, 表示两个GO术语X和Y之间的相似性。
(1)
(2)
(3)
在给定的PPI网络构成的图 中,构建一个由0/1组成的邻接矩阵A。如果两个蛋白质之间存在相互作用,则用 的值为1,否则 的值为0。根据公式(1)~(3)分别计算矩阵A中, 值为0对应的蛋白质对在BP、CC、CF这三个生物过程中的GO语义相似值 , , 。根据公式(4),计算更新蛋白质对(i,j)的新权值,加入到E中。
(4)
2.2. 基于序列和拓扑信息的核–附属的复合物识别算法
基于复合物的特点,本文在拓扑方面利用子图的密度和直径;在生物特征方面,结合本研究的前期工作 [13] ,引入氨基酸背景频率的生物学特征作为生物学特征,具体描述如(5)~(7)。
1) 密度: 是有权图中节点v的度, 是网络中的边e的权值, 为有权网络中节点的个数。
(5)
2) 子图直径:指网络中任意两节点间距离的最大值。
3) 氨基酸背景频率:
表示组成蛋白质的某个氨基酸, 表示氨基酸 出现在蛋白质构成的子图中的次数, 表示子图中包含蛋白质序列长度之和, 表示子图中蛋白质的个数。 表示子图中的某一个蛋白质 的序列长度。
(6)
4) 余玄相似度:
利用余弦相似性描述两个蛋白质之间的相似程度,其中n = 20,表示组成蛋白质的20个氨基酸在某一个蛋白质中出现的频率用向量 表示, 和 分别表示蛋白质i对应向量 和蛋白质j对应的向量 向量中的第i和第j分量值。
(7)
核心-附属物结构是蛋白质复合物重要结构特征之一。MWCA算法在聚类步骤中用两步先后挖掘网络中可能组成蛋白质复合物的核心蛋白质和附属蛋白质。首先通过前面构建的有权网络模型中选择度最大的节点作为初始簇,利用条件(8)将该簇周围满足扩充条件的蛋白质节点扩展构成核心簇,然后在剩余的节点中重复前面的操作,直到全部遍历网络节点,最后形成核心蛋白质簇集合,其中条件(8)中基于最优策略, , , 。其次,将识别到的蛋白质核心簇映射到DIP网络中,通过合并策略添加附属簇。
(8)
算法:MWCA
输入:PPI网络,GO数据,
输出:识别的复合物集合CL
第一步:核心蛋白质复合物识别
1) 对于初始的无权蛋白质网络,集合GO信息
2) For 计算两个蛋白质对应注释集合GO的相似度
3) 计算网络中蛋白质节点之间语义相似度
4) end for
5) 根据语义相似度构建有权网络模型G
6) 初始化蛋白质复合物集合DS
7) For有权网络模型G
8) 选择G中最大度的节点V作为初始簇,
9) 将V周边的节点利用条件(8)扩展加入初始簇中
10) G = G − V
11) GO TO 8),直到G为空。
12) 得到核心簇集合
第二步:
1) 将每个核心蛋白质复合物 映射到DIP蛋白质相互作用网络GD中得 ;
2) 对于每一对 ,如果 与 之间的 (t = 0.2)
complex ( ) = core( ) U attachment( ),输出识别的蛋白质簇 。
3) 输出识别的蛋白质复合物集合
3. 实验与结果分析
3.1. 数据集及评价标准
为了验证MWCA算法的有效性,算法将基于酵母菌PPI网络中在两个不同的蛋白质复合物数据集上进行实验,这两个蛋白质复合物数据集来自于CYC2008和MIPS。在计算蛋白质节点之间的语义相似性时采用基于GO数据库注释网络中的蛋白质。
在复合物识别的评测实验中,为了准确评测算法的实验结果,采用recall,precision和f-measure为评价指标。给定一组已知的蛋白质复合物集合 和一组算法得到的簇集合 ,如果定位到的簇 与真实蛋白质模块 满足条件 ,则认定 和 是匹配的。 是识别的蛋白质模块 和真实蛋白质模块 之间交集的大小, 和 分别为检测到的复合物大小和真实的复合物的大小。
(9)
(10)
(11)
(12)
3.2. 实验结果分析
为了分析MWCA算法的性能,实验将MWCA与3个经典蛋白质复合物算法进行比较,即CFinder,MCode和MCL算法。其中CFinder和MCode是典型的基于密度的复合物预测方法。MCL是基于流模拟的复合物预测算法。本文实验中,3种方法的参数均采用文献中的默认参数值 [5] [6] [7] ,实验结果对比如图1和图2。
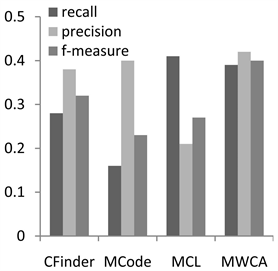
Figure 1. Performance comparison on CYC2008
图1. 在CYC2008上性能比较
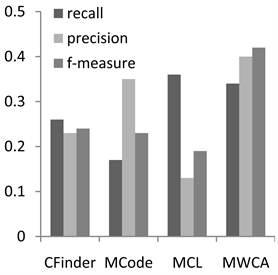
Figure 2. Performance comparison on MIPS
图2. 在MIPS上性能比较
图1为CYC2008数据集上的实验结果,本算法能够在综合性的评价指标F值上取得较好的效果。在召回率方面优于CFinder和MCode,仅次于MCL。在准确率方面,MWCA算法最优,这说明结合基因本体和序列信息可以有效地降低PPI的噪声数据对识别的复合物数量的影响,提高与真实复合物匹配的预测复合物的数量。在F值评价结果可以看出,MWCA取得了比较好的结果。从两个图中我们可以看出在MIPS上的评价结果与在CYC2008上类似,我们的方法和其他方法相比在准确率和F值能达到更高的值。
3.3. 不同阈值t条件下算法性能比较
为了进一步说明MWCA算法的有效性,图3和图4列出了四种算法在9个不同阈值 条件下F值的表现性能,算法整体的性能要优于其他三类算法。
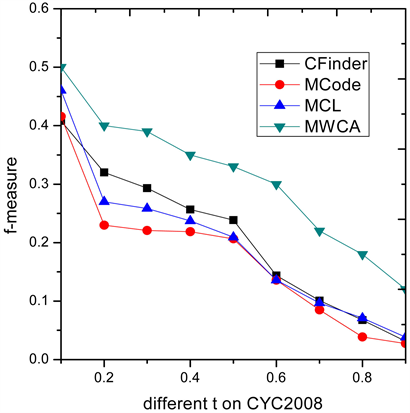
Figure 3. F-measure comparison on CYC2008
图3. 在CYC2008上F-measure性能比较
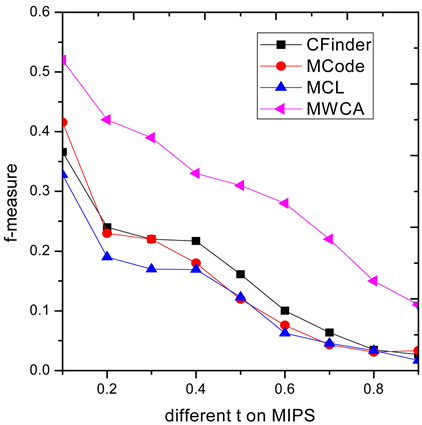
Figure 4. F-measure comparison on MIPS
图4. 在MIPS上F-measure性能比较
3.4. 功能性分析
对于本文提出的算法MWCA,除了前面从量上进行对比和分析,本节将从功能性上进行比较和分析,即Pvalue值。识别到的蛋白质簇对于某一生物功能的富集程度通常用此值进行分析,进而评测算法识别到的复合物的有效性 [14] 。Pvalue值得计算基于GO注释,如(13)。
(13)
其中, 表示PPI网络中所包含的蛋白质节点的个数, 表示算法识别到某个复合物C中蛋白质个数,l代表识别的蛋白质复合物C中含有某个功能F的蛋白质数量, 表示蛋白质网络中含有该功能的蛋白质个数。通常具有相近功能的蛋白质处于相同的复合物内。如果预测的蛋白质复合物具有较小Pvalue值,则说明预测算法识别到的蛋白质复合物出现这类生物功能的概率就越高,因而预测蛋白质复合物正确的可能性就越大。一般情况下,Pvalue值小于0.001的预测的复合物是有实际意义的。表1列出了该方法所识别的部分复合物的功能注释。

Table 1. Comparison of functional enrichment
表1. 在CYC2008中检测到的部分蛋白质复合物功能注释
4. 结论
蛋白质复合物由拓扑特征和生物特性共同决定。只采用某一特征进行复合物识别势必会影响识别的整体性能。本文提出一种新的融合多源信息的复合物识别算法,融合了拓扑特征和生物信息识别蛋白质复合物,以有效的识别PPI网络中具有复杂结构特征的复合物。本文基于GO的语义相似性构建有权的蛋白质网络模型,设计以稠密子图的密度、直径和余玄相似度为聚类条件的核–附属的聚类算法,根据节点度的值为种子节点进行聚类,在加权蛋白质网络中将种子节点不断聚类扩展识别蛋白质复合物。为验证MWCA的有效性,本文提出的算法与3个典型的复合物识别算法进行比较和分析可以看出算法在准确率,F度量和功能富集分析都有了较大的性能提升。
基金项目
辽宁省教育厅科技项目(L201605)和辽宁省自然科学基金项目(201802068)。
文章引用
于杨. 一种新的基于多源信息的复合物识别算法
A New Method for Protein Complex Detection Based on Multi-Source Information[J]. 生物物理学, 2018, 06(04): 67-75. https://doi.org/10.12677/BIPHY.2018.6464007
参考文献
- 1. 冀俊忠, 刘志军, 刘红欣, 刘椿年. 蛋白质相互作用网络功能模块检测的研究综述[J]. 自动化学报, 2014, 40(4): 577-593.
- 2. 丁玉连, 雷秀娟, 代才. 模拟鸽子优化过程的蛋白质复合物识别算[J]. 计算机科学与探索, 2017, 11(8): 1279-1287.
- 3. Wang, Y. and Qian, X. (2004) Functional Module Identification in Protein Interaction Networks by Interaction Patterns. Bioinformatics, 30, 81-93.
- 4. 于杨. 蛋白质相互作用网络中复合物识别算法研究[D]: [博士学位论文]. 哈尔滨: 哈尔滨工业大学, 2014.
- 5. Bader, G.D. and Hogue, C.W. (2003) An Automated Method for Finding Molecular Complexes in Large Protein Interaction Networks. BMC Bioinformatics, 4, 2. https://doi.org/10.1186/1471-2105-4-2
- 6. Adamcsek, B., Palla, G., Farkas, I.J., Derényi, I. and Vicsek, T. (2006) Cfinder: Locating Cliques and Overlapping Modules in Biological Networks. Bioinformatics, 22, 1021-1023. https://doi.org/10.1093/bioinformatics/btl039
- 7. Van Dongen, S. (2000) Graph Clustering by Flow Simulation. PhD Thesis, University of Utrecht, Utrecht.
- 8. Ahn, Y.Y., Bagrow, J.P. and Lehmann, S. (2010) Link Communities Reveal Multiscale Complexity in Networks. Nature, 466, 761-764. https://doi.org/10.1038/nature09182
- 9. Wang, J., Zheng, W., Qian, Y. and Liang, J. (2017) A Seed Expansion Graph Clustering Method for Protein Complexes Detection in Protein Interaction Networks. Molecules, 22, 2179. https://doi.org/10.3390/molecules22122179
- 10. Lakizadeh, A. and Jalili, S. (2016) BICAMWI: A Genetic-Based Biclustering Algorithm for Detecting Dynamic Protein Complexes. PLOS One, 11, e0159923. https://doi.org/10.1371/journal.pone.0159923
- 11. Xu, Y., Zhou, J., Zhou, S. and Guan, J. (2017) Cpredictor3.0: Detecting Protein Complexes from PPI Networks with Expression Data and Functional Annotations. BMC Systems Biology, 11, 135. https://doi.org/10.1186/s12918-017-0504-3
- 12. 汤希玮, 李勇帆, 胡秋玲. 结合多数据源预测蛋白质复合物[J]. 计算机工程与应用, 2012, 48(27): 105-108.
- 13. Yang, Y., Liu, J., Feng, N., Song, B. and Zheng, Z. (2017) Combining Sequence and Gene Ontology for Protein Module Detection in the Weighted Network. Journal of Theoretical Biology, 412, 107-112. https://doi.org/10.1016/j.jtbi.2016.10.010
- 14. Vlaic, S., Conrad, T., Tokarskischnelle, C., Gustafsson, M., Dahmen, U., Guthke, R. and Schuster, S. (2018) Modulediscoverer: Identification of Regulatory Modules in Protein-Protein Interaction Networks. Scientific Reportsvolume, 8, Article Number: 433. https://doi.org/10.1038/s41598-017-18370-2