Journal of Organic Chemistry Research
Vol.
12
No.
01
(
2024
), Article ID:
83620
,
16
pages
10.12677/jocr.2024.121010
烯烃的不对称官能化反应研究 进展
曾宣云
兰州交通大学,化学与化工学院,甘肃 兰州
收稿日期:2024年2月20日;录用日期:2024年3月21日;发布日期:2024年3月29日

摘要
由于C=C键的存在,烯烃被认为是现代有机化学中最有吸引力的底物之一。利用金属催化剂和有机催化剂在烯烃上催化加成H-Y (Y = H, B, C, N, Si等)是生成新的C-H键和C-Y键最常用和最有效的方法之一。不对称烯烃的氢官能化提供了一个直接的策略,以获得对映异构富集的分子。本文综述了利用金属和有机催化剂催化烯烃的不对称加氢官能化反应研究的进展。
关键词
烯烃,金属催化,有机催化,不对称官能化

Research Progress in Asymmetric Functionalization of Olefins
Xuanyun Zeng
Department of Chemistry and Chemical Engineering, Lanzhou Jiaotong University, Lanzhou Gansu
Received: Feb. 20th, 2024; accepted: Mar. 21st, 2024; published: Mar. 29th, 2024

ABSTRACT
Olefins are considered to be one of the most attractive substrates in modern organic chemistry due to the existence of the C=C bond. The catalytic addition of H-Y (Y = H, B, C, N, Si, etc.) on olefins using metal catalysts and organic catalysts is one of the most commonly used and effective methods to generate new C-H bonds and C-Y bonds. Hydrogen functionalization of asymmetric olefins provides a direct strategy to obtain enantioenriched molecules. In this paper, the research progress of asymmetric hydrogenation functionalization of olefins catalyzed by metal and organic catalysts is reviewed.
Keywords:Olefins; Metal Catalysis, Organic Catalysis, Asymmetric Functionalization

Copyright © 2024 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/


1. 引言
烯烃的催化不对称加氢官能化可以从简单的化学原料中直接获得一系列对映体富集的手性骨架,这些活性骨架广泛应用于众多领域,因此,烯烃的不对称官能化备受关注。近几十年来,均相不对称烯烃加氢官能化催化领域主要以贵金属催化为主 [1] 。大部分的贵金属都是地壳中的稀有元素,价格昂贵。此外,贵金属的毒性造成了严重的问题,特别是在制药业。考虑到成本问题,地球上丰富的过渡金属可以成为稀有贵重金属的极好替代品 [2] 。与贵金属相比,地球上储量丰富的过渡金属与烯烃的配位能力较弱,通常经历一次电子氧化还原循环。从绿色和可持续的角度,又发展出有机催化,比起金属催化,有机催化仅适用于结构特殊的烯烃底物,需合理设计底物与催化剂,适用范围有限 [3] 。本文将从以下几类对烯烃的不对称官能化进行综述,分别为氢胺化、氢芳基化、氢硅烷化、氢甲酰化、氢烷氧化、Friedel-Crafts烷基化。
2. 烯烃的不对称官能化
2.1. 氢胺化
氢胺化反应是一种通过在烯烃上加成胺而直接形成C−N键的反应,是一种在精细化学,药品,农用化学品,溶剂和表面活性剂中获得胺的途径。
Buchwald课题组 [4] 报道了通过CuH催化1-取代环丁烯和环丙烯的氢胺化反应,各种类型的多取代氨基环丁烷和氨基环丙烷的非对映和对映选择性合成。并通过DFT计算研究了张力三取代烯烃反应活性增强的原因,以及与环丁烯和环丙烯反应的区域选择性和对映选择性的差异。(见图1)
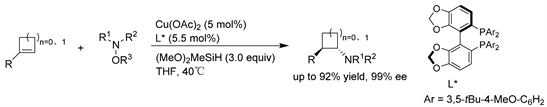
Figure 1. Cu-catalyzed asymmetric hydroamination of cyclobutene and cyclopropene
图1. Cu催化的环丁烯和环丙烯的不对称氢胺化
Buchwald课题组 [5] 又报道了一种采用铜催化的不对称氢胺化反应合成手性叔胺的温和方法。取代以区域选择性的方式发生,在苯乙烯衍生物的α位置生成C-N键。该方法已被证明与各种取代苯乙烯衍生物和β取代苯乙烯相容。(见图2)

Figure 2. Cu-catalyzed asymmetric hydroamination of styrene derivatives
图2. Cu催化的苯乙烯衍生物的不对称氢胺化
Buchwald课题组 [6] 开发了第一个对映选择性铑催化的非活化末端烯烃分子内氢胺化反应。利用二萘基富电子膦配体,以高达91%的ee值合成了多种富集对映体的2-甲基吡咯烷。(见图3)

Figure 3. Rh-catalyzed asymmetric intramolecular hydroamination of unactivated terminal alkenes
图3. Rh催化的非活化端烯的不对称分子内氢胺化
Hannedouche课题组 [7] 报道了由一系列手性N-苄基取代联萘二胺原位制备手性酰胺烷基酸钇配合物。这些手性杂配体配合物被证明是在高反应温度下对位阻较大的烯烃进行不对称分子内氢胺化反应的有效催化剂。对它们的手性环境进行微调,可以使含有1,2-二烷基取代的碳-碳双键的氨基烯烃的环化反应达到高达77%的ee值,这是首次报道通过氢胺化反应形成对映富集的季碳中心。(见图4)
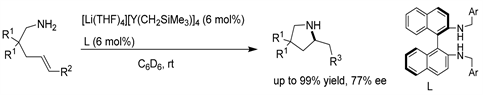
Figure 4. Y-catalyzed asymmetric intramolecular hydroamination of aminoolefins
图4. Y催化的氨基烯烃不对称分子内氢胺化
Hartwig课题组 [8] 还报道了未活化α-烯烃和双环烯烃与芳酰胺和磺胺的分子间胺化反应,并确定了催化剂的静息状态和添加烯烃的中间体中的成分。该方法包括用Ir催化双环烯烃与酰胺和磺胺形成高收率和高ee产品的反应。(见图5)

Figure 5. Ir-catalyzed asymmetric hydroamination of norbornene derivatives
图5. Ir催化的降冰片烯衍生物的不对称氢胺化
Liu课题组 [9] 报道了第一个具有高度对映选择性的Brønsted酸催化的烯烃分子内氢胺化反应,能够高效地构建一系列具有α-四取代碳立体中心的手性(螺环)吡咯烷(ee值高达96%),具有优异的官能团耐受性,反应底物的范围相当广泛。该策略的一个独特之处在于利用硫脲基团作为激活基团和导向基团,通过与Brønsted酸和双键的合作多重氢键形成。该方法的实用性突出体现在易于构建手性合成中间体和重要的结构骨架,这些骨架在有机合成中广泛存在。(见图6)

Figure 6. Brønsted acid-catalyzed asymmetric intramolecular hydroamination of olefins
图6. Brønsted酸催化的烯烃的不对称分子内氢胺化
Beauchemin课题组 [10] 报道了一种催化偶联策略,简单的醛预催化剂通过暂时的分子内反应,使室温下分子间的氢胺化反应和邻二胺的合成成为可能。该催化剂允许烯丙基胺和羟胺形成混合胺,以促进分子内的氢胺化反应。用手性醛获得的对映体选择性也突出了这种催化偶联策略在不对称催化中的潜力,并证明了仅依靠临时的分子内偶联进行高效的对映体诱导是可能的。(见图7)
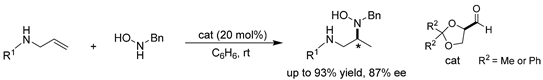
Figure 7. Aldehyde-catalyzed asymmetric hydroamination of allylamines
图7. 醛催化的烯丙基胺的不对称氢胺化
Hultzsch课题组 [11] 报道了利用手性稀土金属铱催化剂将简单胺立体选择性加成到非活化烯烃上,以中等产率和对映选择性(最高可达61% ee)获得目标氢胺化产物。苄胺因其苄基可以很容易地去除,相应的手性伯2-氨基烷烃可以直接从1-烯烃中得到。(见图8)
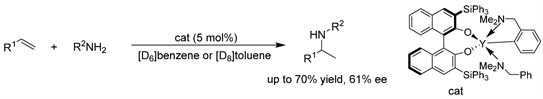
Figure 8. Y-catalyzed asymmetric hydroamination of unactivated terminal alkenes
图8. Y催化的非活化端烯的不对称氢胺化
Buchwald课题组 [12] 开发了一种铜催化烯烃与芳胺邻苯甲酸酯类化合物的氢胺化反应,用于制备对映体富集的叔芳胺。tBuOH的使用,以及催化量的PPh3,对于使用N-芳基羟胺酯作为亲电性氮源是至关重要的。该方法成功地应用于从各种烯烃类底物中合成具有多种官能团的α和β-手性芳胺。(见图9)
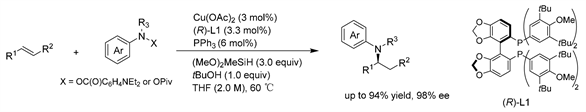
Figure 9. Cu-catalyzed asymmetric hydroamination of olefins
图9. Cu催化的烯烃的不对称氢胺化
2.2. 氢芳基化
Pedro课题组 [13] 报道了羟基吲哚与硝基烯烃的不对称催化反应,实现了硝基烯烃的氢芳基化,对映选择性合成含碳环取代吲哚的2-氨基1,1-二芳烷。本文描述的不对称催化反应是在吲哚的碳环上以硝基烯烃为亲电试剂的第一个区域和对映选择性烷基化反应,该反应是由市售的Rawal squaramide催化的。在我们的反应条件下,羟基吲哚在碳环上反应,而不是在唑体系中反应,因为羟基既是激活基团又是导向基团。该方法基于吲哚碳环上羟基的活化/导向作用,以较高的产率、区域选择性和对映选择性得到C-4、C-5和C-7位官能团化的硝基烷基化吲哚。(见图10)
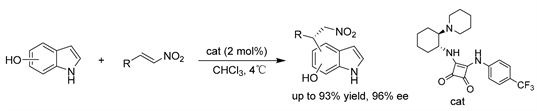
Figure 10. Squaramide-catalyzed asymmetric hydroarylation of nitroolefins
图10. 方酰胺催化的硝基烯烃的不对称氢芳基化
Pedro课题组 [14] 还报道了使用市售的手性方酰胺催化硝基炔烯与4-羟基吲哚不对称氢芳基化的反应方案。通过对反应条件的优化,得到了产率良好,对映选择性极佳(高达97% ee)的C5位烷基化吲哚,并以13:3:1的比例得到了C5-C7二烷基化副产物和C7烷基化副产物。(见图11)

Figure 11. Squaramide-catalyzed asymmetric hydroarylation of nitroalkynes
图11. 方酰胺催化的硝基炔烯的不对称氢芳基化
赵军岭课题组 [15] 通过在吲哚环的C4位引入烷基胺片段,开发了一种手性磷酸催化的β,γ-不饱和α-酮亚胺酯在吲哚C7位高度区域和对映选择性的不对称氢芳基化反应。该方法适用于广泛的4-氨基吲哚和β, γ-不饱和α-酮亚胺酯,可获得相应的C7位功能化手性吲哚衍生物,收率高(高达97%),对映选择性中等至优异(最高至99% ee)。(见图12)
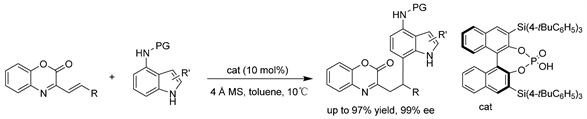
Figure 12. CPA-catalyzed asymmetric hydroarylation of indole with β, γ-unsaturated α-ketoimine esters
图12. 手性磷酸(CPA)催化的吲哚和β, γ-不饱和α-酮亚胺酯的不对称氢芳基化
赵军岭课题组 [16] 又报道了2-丁烯-1,4-二酮或3-芳酰基丙烯酸酯与4-氨基吲哚进行对映选择性氢芳基化反应的有效方案。该反应由一种手性磷酸催化,得到相应的1,4-二取代吲哚,产率中高(高达95%),对映选择性良好(高达88% ee),反应可在不损失效率的情况下以克为单位进行。(见图13)
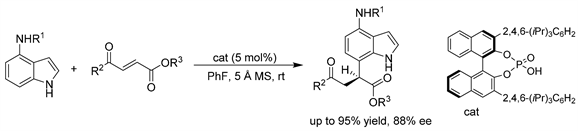
Figure 13. CPA-catalyzed asymmetric hydroarylation of indoles with 3-aroyl acrylates
图13. 手性磷酸催化的吲哚与3-芳酰基丙烯酸酯的不对称氢芳基化
Ellman课题组 [17] 利用Rh/手性磷酰胺催化剂体系,通过定向C-H键激活,开发了一种高度立体选择性的烯烃分子内氢芳基化反应,这是一个少见的涉及芳香C-H键激活的对映选择性催化反应的例子。含有1,1-和1,2二取代烯烃以及三取代烯烃的底物均实现了环化,每种底物类别产物的对映选择性>90% ee。含有Z-烯烃异构体的底物比含有E-烯烃异构体的底物更有效地进行环化。这种立体选择性催化转化提供了一系列具有不同取代模式的手性茚、二氢苯并呋喃和二氢吡咯吲哚,因此应适用于一系列生物活性化合物的不对称合成。(见图14)
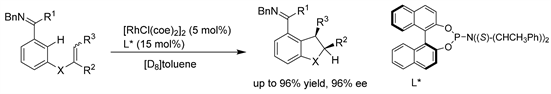
Figure 14. Rh-catalyzed asymmetric intramolecular hydroarylation of olefins
图14. Rh催化的烯烃的不对称分子内氢芳基化
Cavallo课题组 [18] 报道了一种使用铱(I)/喹氧磷催化剂体系进行非活化烯烃对映选择性氢化芳基化的通用高效方案。该反应在温和的条件下进行,具有较高的官能团耐受性,可从稳定、易于处理的原料中快速构建具有重要生物学意义的手性二氢苯并呋喃,具有良好的产率和对映选择性。简单的芳香族酮或酰胺作为导向基团,允许在更阻碍的邻位上进行区域选择反应。叔苄基立体中心在温和的反应条件下,可以以完全的原子经济性从现成的原料中直接得到。(见图15)
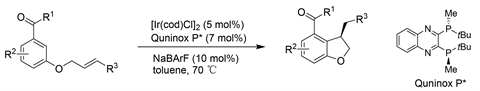
Figure 15. Ir-catalyzed asymmetric intramolecular hydroarylation of olefins
图15. Ir催化的烯烃的不对称分子内氢芳基化
2.3. 氢硅烷化
硅碳键形成反应,包括C-H/C-X硅烷化反应、硅氢加成反应和Si-C键的交叉交换,一直被认为是有机硅化学的关键课题和基石,在有机合成和功能材料方面具有重要价值。
Nishiyama课题组 [19] 报道了手性醋酸铑配合物与双(恶唑啉基)苯基配体催化芳基烯烃的高效、高对映选择性硅氢加成反应,得到了ee值高达99%的手性硅烷衍生物。这种催化剂显著提高了催化活性和产物的对映选择性,并发现反式和顺式β-甲基苯乙烯的硅氢化反应产生相同的对映体,具有区域选择性和对映选择性。(见图16)
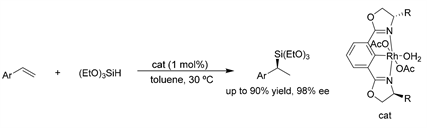
Figure 16. Rh-catalyzed asymmetric hydrosilylation of styrene derivatives
图16. Rh催化的苯乙烯衍生物的不对称氢硅烷化
Lu课题组 [20] 开发了一种高对映选择性的钴催化烯烃与硅烷的马尔科夫尼科夫型硅氢化反应,以构建更有价值的手性二氢硅烷。该反应操作简单,原子经济,具有良好的官能团耐受性。对于乙烯基芳烃,手性苯基硅烷的平均产率为82%,ee值优于98%。在优化条件下,脂肪族烯烃也可在81-87% ee值范围内制备手性脂肪族二氢硅烷。(见图17)

Figure 17. Co-catalyzed asymmetric hydrosilylation of alkenes
图17. Co催化的烯烃的不对称氢硅烷化
Xu课题组 [21] 报道了一种高对映选择性的Si-C偶联反应,通过使用钯催化剂与手性TADDOL衍生的亚磷酰胺配体对羰基活化的烯烃进行硅氢加成反应,从而抑制了O-硅氢加成/烯烃还原。通过对马来酰亚胺的立体专一性Si-C偶联/硅氢加成反应,以高达99%的产率、>99:1的非对映选择性和>99:1的对映选择性得到了一系列的硅基琥珀酰亚胺类。高度的立体选择性可以远程控制轴向手性,从而产生功能化的轴向手性琥珀酰亚胺,通过对映选择性构建多达3个立体中心的氮杂环,突出了产物的实用性。(见图18)
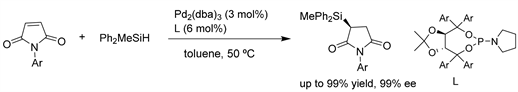
Figure 18. Pd-catalyzed asymmetric hydrosilylation of carbonyl activated olefins
图18. Pd催化的羰基活化烯烃的不对称氢硅烷化
Xu课题组 [22] 又报道了手性铑催化剂促进1,1-二取代环丙烯的高对映选择性非对称氢硅烷化反应。该方法以100%原子效率的方式从现成的起始环丙烯和硅烷中直接获得具有潜在价值的季碳立体中心的手性有机硅化合物,收率高,具有良好的非对映和对映选择性(高达>99:1 dr和>99% ee)。铑催化的过程展示了具有季碳立体中心的手性三元碳环的不对称构建的广泛范围和显著的配体导向的环丙烯高对映选择性硅氢加成反应过程,这也为通过Si-C成键反应制备具有各种取代基的手性硅基环丙烷提供了一种温和而便捷的途径。(见图19)
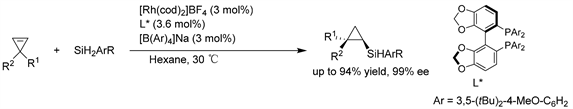
Figure 19. Rh-catalyzed asymmetric hydrosilylation of disubstituted cyclopropenes
图19. Rh催化的二取代环丙烯的不对称氢硅烷化
2.4. 氢甲酰化
烯烃氢甲酰化反应是工业上均相催化剂催化的重要反应之一。光学活性醛是各种药物,农用化学品和其他精细化学品的多功能中间体,区域和立体选择性氢甲酰化已成为合成醛类物质中一个有吸引力的工具。
Morken课题组 [23] 开发了一种铑催化的对映选择性氢甲酰化,它采用了现成的Ph-BPE配体,催化1-烯烃的区域选择性不对称氢甲酰化反应,并迅速生成α-手性醛。这种方法能够从简单的烯烃中有效地合成许多重要和有用的手性基序。广泛的末端烯烃底物适用于该反应,提供了具有良好的对映体选择性和中等至良好的区域选择性的氢甲酰化产物。(见图20)
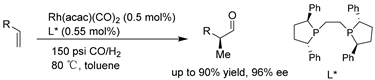
Figure 20. Rh/Ph-BPE-catalyzed asymmetric hydroformylation of terminal olefins
图20. Rh/Ph-BPE催化的末端烯烃的不对称氢甲酰化
Landis课题组 [24] 报道了铑催化的末端和内部芳烯烃与二氮磷烷配体的不对称氢甲酰化。在优化的反应条件下,对大多数底物获得了高的对映选择性(>90% ee)和区域选择性。对于末端烯烃,对映体选择性和区域选择性都与一氧化碳分压成正比,但与氢压力无关。对取代苯乙烯衍生物的氢甲酰化对含有吸电子取代基的底物具有最高的区域选择性。(见图21)

Figure 21. Rh/Bisdiazaphos-catalyzed asymmetric hydroformylation of terminal and internal olefins
图21. Rh/Bisdiazaphos催化的末端烯烃和内烯烃的不对称氢甲酰化
Zhang课题组 [25] 报道了空气稳定和可调的手性双二氢苯并氧膦配体(BIBOPs)用于铑催化的各种末端烯烃的不对称氢甲酰化反应,具有优异的转化率(>99%),中等至优异的对映选择性(高达95:5 er)。易于合成和良好的空气稳定性使BIBOP配体在氢甲酰化反应领域中作为一种独特的配体结构脱颖而出。(见图22)
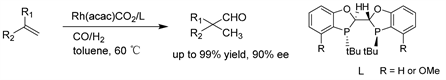
Figure 22. Rh/BIBOPs-catalyzed asymmetric hydroformylation of terminal olefins
图22. Rh/BIBOPs催化的末端烯烃的不对称氢甲酰化
Reek课题组 [26] 描述了在铑催化的二氢呋喃和吡啶底物的不对称氢甲酰化反应中杂化配体的设计和应用,产物具有高水平的区域和对映体选择性。通过N-(叔丁氧羰基)-3-吡咯啉衍生的3-甲醛产物的不对称合成,证明了小杂环烯烃选择性不对称氢甲酰化的实用性,该产物随后转化为氨基酸β-脯氨酸,提供了生成这一重要手性基序的第一个催化方案。(见图23)
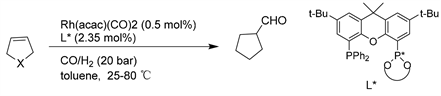
Figure 23. Rh-catalyzed asymmetric hydroformylation of dihydrofuran and pyridine
图23. Rh催化的二氢呋喃和吡啶的不对称氢甲酰化
Zhang课题组 [27] 研究发现,当(S, R)-Bn-Yanphos配体用于Rh催化的乙烯基杂环芳烃和烯丙基-1H-吲哚的不对称氢甲酰化反应时,表现出优异的催化活性,以较高的区域、对映选择性及较好的收率得到手性α-杂环芳醛(高达96% ee)。(见图24)
Reider课题组 [28] 报道了在温和条件下,Rh(CO)2(acac)/TangPhos催化体系是降冰片烯不对称氢甲酰化反应的高对映选择性催化剂。将该方案应用于其他[2.2.1]-双环烯烃的去对称反应,得到中等至优异的对映选择性(55~92% ee)。(见图25)

Figure 24. Rh-catalyzed asymmetric hydroformylation of heterocyclic aromatic hydrocarbons
图24. Rh催化的乙烯基杂环芳烃的不对称氢甲酰化

Figure 25. Rh-catalyzed asymmetric hydroformylation of norbornene derivatives
图25. Rh催化的降冰片烯衍生物的不对称氢甲酰化
2.5. 氢烷氧化
烯烃的氢烷氧化被定义为在C-C双键上加成一个羟基基团,这为从现成的烯烃合成价值更高的醚提供了一种原子经济和非常直接的方法。
Li课题组 [29] 设计、合成了一系列手性1,1′-联萘-2,2′-二磺酸,并将其应用于Ti催化的非活化烯烃的高对映选择性分子内氢烷氧化环化反应。配体的磺酰胺基团和水是催化所必需的,因为它们可以通过氢键稳定具有催化活性的配合物。在温和的条件下,以高达99%的产率和高达97%的对映体过量值获得了各种2-甲基香豆素。(见图26)
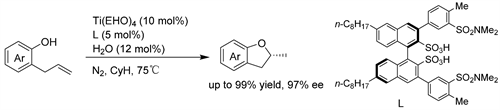
Figure 26. Ti/naphthalenesulfonic acid-catalyzed asymmetric intramolecular hydroalkane oxidation of unactivated olefins
图26. Ti/联萘磺酸催化的非活化烯烃的不对称分子内氢烷氧化
Hintermann课题组 [30] 通过2-烯丙基苯酚环异构化生成2-甲基-2,3-二氢苯并呋喃(2-甲基香豆素),实现了醇(酚)与非活化烯烃的不对称氢烷氧化反应。手性钛-羧酸配合物在不对称催化反应的罕见高温下催化了该反应。催化剂由异丙醇钛、手性配体(aS)-1-(2-甲氧基-1-萘基)-2-萘酸或其衍生物和共催化量的水按1:1:1的比例(各5mol %)混合生成。这种均相热催化(HOT-CAT)在240℃下,以高达90%的产率和高达85%的ee值得到各种(S)-2-甲基香豆素。(见图27)
Zhao课题组 [31] 开发了一种以手性磷酰胺和TiCl4组合为催化剂的非活化烯烃不对称分子内氢烷氧化环化新方法,制备了多种手性四氢呋喃衍生物。尽管对映选择性适中,但这项工作为具有挑战性的未活化烯烃的不对称氢烷氧基化提供了一种新的催化策略。(见图28)
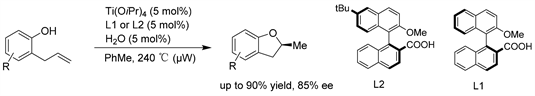
Figure 27. Ti/binaphthyl carboxylic acid-catalyzed asymmetric intramolecular hydroalkane oxidation of unactivated olefins
图27. Ti/联萘羧酸催化的非活化烯烃的不对称分子内氢烷氧化

Figure 28. Ti/binaphthylphosphoramide-catalyzed asymmetric intramolecular hydroalkane oxidation of unactivated olefins
图28. Ti/联萘磷酰胺催化的非活化烯烃的不对称分子内氢烷氧化
List课题组 [32] 通过高度受限和高度酸性的Brønsted酸实现电子和空间无偏烯烃的不对称分子内氢烷氧基化,快速获得具有生物活性的1,1-二取代四氢呋喃。该方法操作简单,可以以较高的产率和对映选择性转化多种烯醇。该方法在烯烃的各种其他氢官能化和相关反应中具有巨大的潜力。(见图29)

Figure 29. IDPi-catalyzed asymmetric intramolecular hydroalkoxylation of enols
图29. 亚氨基双磷酰亚氨酸酯(IDPi)催化的烯醇的不对称分子内氢烷氧基化
List课题组 [33] 报道了基于C2对称的磷酰亚胺酸骨架的受限Brønsted酸的设计和合成,实现了催化羟基烯醇醚的对映选择性螺旋缩醛化反应,双环螺缩酮类产物产率高达88%,ee值高达97%。这些合理构建的Brønsted酸具有极其苛刻空间要求的手性微环境,具有单一的催化相关和几何限制的双功能活性位点。该催化剂设计有望在涉及小型和结构或功能无偏向性底物的催化不对称反应中得到广泛的应用。(见图30)

Figure 30. IDP-catalyzed asymmetric intramolecular hydroalkane oxidation of enol ethers
图30. 二磷酸(IDP)催化的烯醇醚的不对称分子内氢烷氧化
Nagorny课题组 [34] 发现在BINOL衍生的CPA催化剂作用下,烷基侧链上含有叔羟基的环状烯醇醚可以以高达96%的产率和高达96%的ee值形成相应的螺环缩酮。手性催化剂可以克服热力学螺缩酮形成的固有偏好,在反应条件下可以实现非热力学螺缩酮的高选择性形成。(见图31)
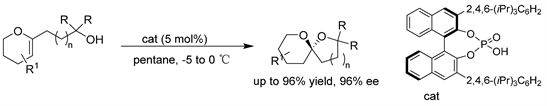
Figure 31. CPA-catalyzed asymmetric intramolecular hydroalkane oxidation of enol ethers
图31. 手性磷酸催化的烯醇醚的不对称分子内氢烷氧化
Hartwig课题组 [35] 报道了由铱-(S)-DTBM-SEGPHOS配合物催化的非活化脂肪族烯烃与酚的分子间不对称氢烷氧化反应。该反应不是单纯的酸催化,相反,它可能是通过还原消除观察到的烯丙基铱氢化物物种生成Ir (I)中间体,随后发生可逆O-H键氧化加成、限制翻转的烯烃插入和产物释放的还原消除。(见图32)
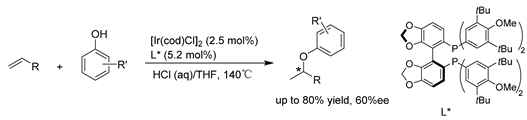
Figure 32. Ir-catalyzed asymmetric hydroalkane oxidation of aliphatic olefins
图32. Ir催化的脂肪族烯烃的不对称氢烷氧化
Maruoka课题组 [36] 报道了八氢联萘二胺类有机小分子催化剂可用于实现醇与α,β-不饱和的首次不对称氢烷基化反应。该体系可以将甲醇、乙醇和烯丙醇加成到脂肪族α,β-不饱和醛中,以中等产率和对映选择性得到目标产物。催化剂中的磺酰胺部分对于亚胺离子的形成是必不可少的。(见图33)

Figure 33. Octahydrobinaphthyldiamine-catalyzed asymmetric hydroalkylation of α, β-unsaturated aldehydes
图33. 八氢联萘二胺催化的α,β-不饱和醛的不对称氢烷基化
2.6. Friedel-Crafts烷基化
烯烃的Friedel-Crafts烷基化反应是生成新的C-C键的有效方法,其中,含功能化吲哚骨架的化合物是最重要的含氮杂环化合物之一,在许多研究领域有广泛的应用,故吲哚的不对称Friedel-Crafts烷基化又备受关注。
He课题组 [37] 通过含手性N-杂环卡宾的NiH配合物催化剂,实现了乙烯基芳烃与末端烯烃的不对称尾接尾Friedel-Crafts烷基化。该反应提供了具有高对映选择性(高达94% ee)和化学选择性(交叉偶联/自偶联产物比:高达99:1)的二取代烯烃。乙烯基芳烃和NHC配体N-芳基上取代基的电子效应,而不是π-π堆积机理,有助于立体效应,影响Friedel-Crafts烷基化的结果。(见图34)
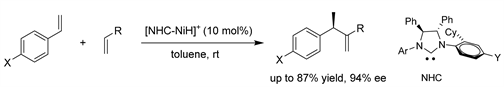
Figure 34. Ni/NHC-catalyzed asymmetric Friedel-Crafts alkylation of terminal olefins
图34. Ni/NHC (氮卡宾)催化的末端烯烃的不对称Friedel-Crafts烷基化
Zhou课题组 [38] 通过使用螺环亚磷酰胺配体开发了一种高效的α-烷基芳基乙烯和乙烯的不对称Friedel-Crafts烷基化的催化体系,并以优异的对映选择性(高达99% ee)构建手性全碳季碳中心提供了一种新的方法。(见图35)
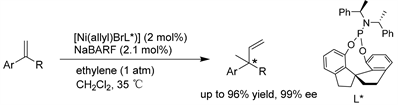
Figure 35. Ni/phosphoramidite-catalyzed asymmetric Friedel-Crafts alkylation of ethylene
图35. Ni/亚磷酰胺催化的乙烯的不对称Friedel-Crafts烷基化
Jaisankar课题组 [39] 利用Fiaud酸(反式-1-羟基-2,5-二苯基磷烷-1-氧化物,一种基于磷烷的磷酸,是高效的手性Brønsted酸),催化吲哚与2-丁烯-1,4-二酮的不对称Friedel-Crafts烷基化反应,在催化剂负载量为10 mol%的情况下,反应顺利进行,得到了2-(吲哚-3-基)丁烷1,4-二酮,产率高(高达82%),对映体选择性高(高达91% ee)。该反应的优点在于反应条件足够温和,底物范围广,适用于合成复杂的目标产物。与广泛使用的BINOL衍生的磷酸相比,使用Fiaud's酸作为催化剂可以对2,5-二芳基磷杂烷骨架的立体和电子特性进行调整。(见图36)

Figure 36. Fiaud acid-catalyzed asymmetric Friedel-Crafts alkylation of indoles
图36. Fiaud酸催化吲哚的不对称Friedel-Crafts烷基化
赵军岭课题组 [40] 开发了3-取代吲哚与β,γ-不饱和α-酮亚胺酯高度对映选择性的C2位Friedel-Crafts烷基化反应,手性磷酸催化剂高效地催化了该反应。以α-酮亚胺酯为基序的C2功能化吲哚衍生物,产率可高达93%,对映选择性可达99% ee以上。(见图37)
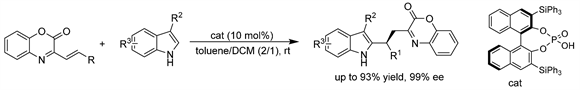
Figure 37. CPA-catalyzed asymmetric Friedel-Crafts alkylation of indoles with β, γ-unsaturated α-ketoimino esters
图37. 手性磷酸催化的吲哚与β, γ-不饱和α-酮亚胺酯的不对称Friedel-Crafts烷基化
Akiyama课题组 [41] 报道了第一个手性磷酸催化吲哚与硝基烯烃的Friedel-Crafts烷基化反应,获得了具有优异对映选择性(高达99% ee)的Friedel-Crafts加成产物。该课题组发现添加活性粉状分子筛3 Å MS显著提高了产物的产率和对映体选择性。(见图38)
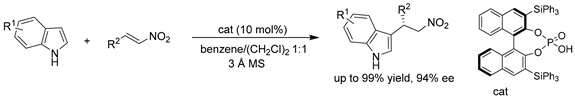
Figure 38. CPA-catalyzed asymmetric Friedel-Crafts alkylation of indoles with nitroalkenes
图38. 手性磷酸催化的吲哚与硝基烯烃的不对称Friedel-Crafts烷基化
Akiyama课题组 [42] 又报道了一种手性磷酸催化吲哚与β-烷氧羰基-β-二取代硝基烯的Friedel-Crafts反应。该反应的范围相当广泛,包括芳基环上带有取代基(不论其电子性质如何)的各种硝基苯乙烯,以及具有不同功能(包括中性、给电子或吸电子基团)的吲哚,都能顺利反应,得到了具有高产率(高达88%)和优异选择性(高达97% ee)的全碳季中心的吲哚。进一步的研究表明,烯烃的几何形状和NH -吲哚衍生物的使用对反应活性和选择性都有影响。(见图39)
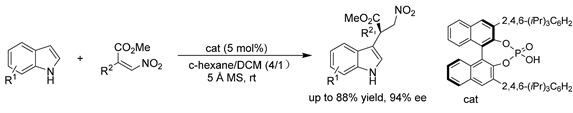
Figure 39. CPA-catalyzed asymmetric Friedel-Crafts reaction of indoles with β-alkoxycarbonyl-β-disubstituted nitroalkenes
图39. 手性磷酸催化的吲哚与β-烷氧羰基-β-二取代硝基烯的不对称Friedel-Crafts反应
3. 总结
近年来烯烃的不对称官能化领域在不断地发展和完善,烯烃的不对称官能团化已成为构建碳–杂键重要有力的策略,本文综述了该领域的研究进展,希望对设计新型催化剂和催化体系提供启发与思路。
文章引用
曾宣云. 烯烃的不对称官能化反应研究进展
Research Progress in Asymmetric Functionalization of Olefins[J]. 有机化学研究, 2024, 12(01): 120-135. https://doi.org/10.12677/jocr.2024.121010
参考文献
- 1. Michon, C., Abadie, M.A., Medina, F. and Agbossou-Niedercorn, F. (2017) Recent Metal-Catalysed Asymmetric Hydroaminations of Alkenes. Journal of Organometallic Chemistry, 847, 13-27. https://doi.org/10.1016/j.jorganchem.2017.03.032
- 2. Du, X. and Zheng, H. (2017) Advances in Base-Metal-Catalyzed Alkene Hydrosilylation. ACS Catalysis, 7, 1227-1243. https://doi.org/10.1021/acscatal.6b02990
- 3. Xie, W.B. and Li, Z. (2020) Asymmetric Synthesis of Ethers by Catalytic Alkene Hydroalkoxylation. Synthesis, 52, 2127-2146. https://doi.org/10.1055/s-0039-1690874
- 4. Feng, S., Hao, H., Buchwald, S.L., et al. (2020) Diastereo-and Enantioselective CuH-Catalyzed Hydroamination of Strained Trisubstituted Alkenes. ACS Catalysis, 10, 282-291. https://doi.org/10.1021/acscatal.9b04871
- 5. Zhu, S.L., Niljianskul, N. and Buchwald, S.L. (2013) Enantio-and Regioselective CuH-Catalyzed Hydroamination of Alkenes. Journal of the American Chemical Society, 135, 15746-15749. https://doi.org/10.1021/ja4092819
- 6. Shen, X.Q. and Buchwald, S.L. (2010) Rhodium-Catalyzed Asymmetric Intramolecular Hydroamination of Unactivated Alkenes. Angewandte Chemie International Edition, 49, 564-567. https://doi.org/10.1002/anie.200905402
- 7. Chapurina, Y., Ibrahim, H. and Hannedouche, J.M. (2011) Catalytic, Enantioselective Intramolecular Hydroamination of Primary Amines Tethered to Di-and Trisubstituted Alkenes. The Journal of Organic Chemistry, 76, 10163-10172. https://doi.org/10.1021/jo202009q
- 8. Sevov, C.S., Zhou, J.R. and Hartwig, J.F. (2012) Iridium-Catalyzed Intermolecular Hydroamination of Unactivated Aliphatic Alkenes with Amides and Sulfonamides. Journal of the American Chemical Society, 134, 11960-11963. https://doi.org/10.1021/ja3052848
- 9. Lin, J.S., Yu, P., Liu, X.Y., et al. (2015) Brønsted Acid Catalyzed Asymmetric Hydroamination of Alkenes: Synthesis of Pyrrolidines BearingaTetrasubstituted Carbon Stereocenter. Angewandte Chemie, 127, 7958-7962. https://doi.org/10.1002/ange.201501762
- 10. MacDonald, M.J., Schipper, D.J. and Beauchemin, A.M. (2011) A Catalytic Tethering Strategy: Simple Aldehydes Catalyze Intermolecular Alkene Hydroaminations. Journal of the American Chemical Society, 133, 20100-20103. https://doi.org/10.1021/ja208867g
- 11. Reznichenko, A.L., Nguyen, H.N. and Hultzsch, K.C. (2010) Asymmetric Intermolecular Hydroamination of Unactivated Alkenes with Simple Amines. Angewandte Chemie International Edition, 49, 8984-8987. https://doi.org/10.1002/anie.201004570
- 12. Ichikawa, S., Zhu, S.L. and Buchwald, S.L. (2018) A Modified System for the Synthesis of Enantioenriched N-Arylamines through Copper-Catalyzed Hydroamination. Angewandte Chemie International Edition, 57, 8714-8718. https://doi.org/10.1002/anie.201803026
- 13. Vila, C., Blay, G., Pedro, J.R., et al. (2018) Enantioselective Synthesis of 2-Amino-1, 1-Diarylalkanes Bearing a Carbocyclic Ring Substituted Indole through Asymmetric Catalytic Reaction of Hydroxyindoles with Nitroalkenes. Organic Chemistry, 83, 6397-6407. https://doi.org/10.1021/acs.joc.8b00612
- 14. Vila, C., Blay, G., Pedro, J.R., et al. (2021) Enantioselective Friedel-Crafts Reaction of Hydroxyarenes with Nitroenynes to Access Chiral Heterocycles via Sequential Catalysis. Organic & Biomolecular Chemistry, 19, 6990-6994. https://doi.org/10.1039/D1OB01238J
- 15. Xun, W., Xu, B., Zhao, J.L., et al. (2018) Regio and Enantioselective Organocatalytic Friedel-Crafts Alkylation of 4-Aminoindoles at the C7-Position. Organic Letters, 20, 590-593. https://doi.org/10.1021/acs.orglett.7b03703
- 16. Huang, T., Zhao, Y., Zhao, J.L., et al. (2019) C7-Functionalization of Indoles via Organocatalytic Enantioselective Friedel-Crafts Alkylation of 4-Amino-Indoles with 2-Butene-1, 4-Diones and 3-Aroylacrylates. Advanced Synthesis & Catalysis, 361, 3632-3638. https://doi.org/10.1002/adsc.201900377
- 17. Ellman, J.A., Harada, H., Thalji, R.K., et al. (2008) Enantioselective Intramolecular Hydroarylation of Alkenes via Directed C-H Bond Activation. The Journal of Organic Chemistry, 73, 6772-6779. https://doi.org/10.1021/jo801098z
- 18. Shinde, V.S., Mane, M.V. and Cavallo, L.G. (2020) Iridium Catalyzed Enantioselective Hydroarylation of Alkenes via C-H Bond Activation: Experiment and Computation. Chemistry: A European Journal, 26, 8308-8313. https://doi.org/10.1002/chem.202001793
- 19. Naito, T., Yoneda, T., Nishiyama, H., et al. (2012) Enantioselective Hydrosilylation of Aromatic Alkenes Catalyzed by Chiral Bis (Oxazolinyl) Phenyl-Rhodium Acetate Complexes. Synlett, 23, 2957-2960. https://doi.org/10.1055/s-0032-1317677
- 20. Cheng, B., Lu, P., Lu, Z., et al. (2017) Highly Enantioselective Cobalt-Catalyzed Hydrosilylation of Alkenes. Journal of the American Chemical Society, 139, 9439-9442. https://doi.org/10.1021/jacs.7b04137
- 21. Gu, X.W., Sun, Y.L., Xu, L.W., et al. (2020)Stereospecific Si-C Coupling and Remote Control of Axial Chirality by Enantioselective Palladiumcatalyzedhydrosilylation of Maleimides. Nature Communications, 11, Article No. 2904. https://doi.org/10.1038/s41467-020-16716-5
- 22. Zhao, Z.Y., Nie, Y.X., Xu, L.W., et al. (2019) Enantioselective Rhodium-Catalyzed DesymmetricHydrosilylation of Cyclopropenes. ACS Catalysis, 9, 9110-9116. https://doi.org/10.1021/acscatal.9b02623
- 23. Morken, J.P., Yu, Z.Y., Eno, M.S., et al. (2015) Enantioselective Hydroformylation of 1-Alkenes with Commercial Ph-BPE Ligand. Organic Letters, 17, 3264-3267. https://doi.org/10.1021/acs.orglett.5b01421
- 24. Watkins, A.L., Hashiguchi, B.G. and Landis, C.R. (2008) Highly Enantioselective Hydroformylation of Aryl Alkenes with Diazaphospholane Ligands. Organic Letters, 10, 4553-4556. https://doi.org/10.1021/ol801723a
- 25. Zhang, X.M., Tan, R.C., Zheng, X., et al. (2016) Tunable P-Chiral Bisdihydrobenzooxaphosphole Ligands for Enantioselective Hydroformylation. Organic Letters, 18, 3346-3349. https://doi.org/10.1021/acs.orglett.6b01452
- 26. Reek, J.N.H., Chikkali, S.H., Bellini, R., et al. (2012) Highly Selective Asymmetric Rh-Catalyzed Hydroformylation of Heterocyclic Olefins. Journal of the American Chemical Society, 134, 6607-6616. https://doi.org/10.1021/ja210117z
- 27. Zhang, X.M., Wei, B., Chen, C.Y., et al. (2017) Efficient Synthesis of (S, R)-Bn-Yanphos and Rh/(S, R)-Bn-Yanphos Catalyzed Asymmetric Hydroformylation of Vinyl Heteroarenes. Organic Chemistry Frontiers, 4, 288-291. https://doi.org/10.1039/C6QO00641H
- 28. Reider P.J., Huang, J.K., Huang, E., et al. (2005) A Highly Enantioselective Catalyst for Asymmetric Hydroformylation of [2.2.1]-Bicyclic Olefins. Tetrahedron Letters, 46, 7831-7834. https://doi.org/10.1016/j.tetlet.2005.09.012
- 29. Xie, W.B. and Li, Z. (2021) Bis(μ-Oxo)—Dititanium(IV)—Chiral Binaphthyldisulfonate Complexes for Highly Enantioselective Intramolecular Hydroalkoxylation of Nonactivated Alkenes. ACS Catalysis, 11, 6270-6275. https://doi.org/10.1021/acscatal.1c01146
- 30. Schlter, J., Blazejak, M., Hintermann, L., et al. (2015) Asymmetric Hydroalkoxylation of Non-Activated Alkenes: Titanium-Catalyzed Cycloisomerization of Allylphenols at High Temperatures. Angewandte Chemie International Edition, 54, 4014-4017. https://doi.org/10.1002/anie.201409252
- 31. Zhao, P.Y., Ma, J.G., Zhao, G.Q., et al. (2019) Asymmetric Intramolecular Hydroalkoxylation of Unactivated Alkenes Catalyzed by Chiral N-Triflyl Phosphoramide and TiCl4. Chinese Journal of Chemistry, 37, 565-569. https://doi.org/10.1002/cjoc.201900544
- 32. List, B., Tsuji, N., Kennemur, J.L., et al. (2018) Activation of Olefins via Asymmetric Brønsted Acid Catalysis. Science, 359, 1501-1505. https://doi.org/10.1126/science.aaq0445
- 33. List, B. and Coric, L. (2012) Asymmetric Spiroacetalizationcatalysed by Confined Brønsted Acids. Nature, 483, 315-317. https://doi.org/10.1038/nature10932
- 34. Nagorny, P., Sun, Z.K., Winschel, G.A. and Borovika, A. (2012) Chiral Phosphoric Acid-Catalyzed Enantioselective and Diastereoselective Spiroketalizations. Journal of the American Chemical Society, 134, 8074-8077. https://doi.org/10.1021/ja302704m
- 35. Sevov, C.S. and Hartwig, J.F. (2013) Iridium-Catalyzed, Intermolecular Hydroetherification of Unactivated Aliphatic Alkenes with Phenols. Journal of the American Chemical Society, 135, 9303-9306. https://doi.org/10.1021/ja4052153
- 36. Maruoka, K.J., Kano, T.C. and Tanaka, Y.H. (2007) Asymmetric Organocatalytic Oxy-Michael Addition of Alcohols to A, B-Unsaturated Aldehydes. Tetrahedron, 63, 8658-8664. https://doi.org/10.1016/j.tet.2007.03.179
- 37. Ho, C.Y., Chan, C.W. and He, L.S. (2015) Catalytic Asymmetric Hydroalkenylation of Vinylarenes: Electronic Effects of Substrates and Chiral N-Heterocyclic Carbene Ligands. Angewandte Chemie International Edition, 54, 4512-4516. https://doi.org/10.1002/anie.201411882
- 38. Shi, W.J., Zhang, Q., Zhou, Q.L., et al. (2006) Highly Enantioselective Hydrovinylation of R-Alkyl Vinylarenes. An Approach to the Construction of All-Carbon Quaternary Stereocenters. Journal of the American Chemical Society, 128, 2780-2781. https://doi.org/10.1021/ja057654y
- 39. Chatterjee, S., Hintermann, L., Jaisankar, P., et al. (2017) Fiaud’s Acid: A Brønsted Acid Catalyst for Enantioselective Friedel-Crafts Alkylation of Indoles with 2-Alkene-1, 4-Diones. Organic Letters, 19, 3426-3429. https://doi.org/10.1021/acs.orglett.7b01383
- 40. Bi, B., Lou, Q.X. and Zhao, J.L. (2015) Chiral Phosphoric Acid Catalyzed Highly Enantioselective Friedel-Crafts Alkylation Reaction of C3-Substituted Indoles to β, γ-Unsaturated α-Ketimino Esters. Organic Letters, 17, 540-543. https://doi.org/10.1021/ol5035222
- 41. Itoh, J., Fuchibe, K. and Akiyama, T. (2008) Chiral Phosphoric Acid Catalyzed Enantioselective Friedel-Crafts Alkylation of Indoles with Nitroalkenes: Cooperative Effect of 3 Å Molecular Sieves. Angewandte Chemie International Edition, 47, 4016-4018. https://doi.org/10.1002/anie.200800770
- 42. Mori, K., Wakazawa, M. and Akiyama, T. (2014) Stereoselective Construction of All-Carbon Quaternary Center by Means of Chiral Phosphoric Acid: Highly Enantioselective Friedel-Crafts Reaction of Indoles with β, β-Disubstituted Nitroalkenes. Chemical Science, 5, 1799-1803. https://doi.org/10.1039/C3SC53542H