Statistics and Application
Vol.
08
No.
01
(
2019
), Article ID:
28726
,
13
pages
10.12677/SA.2019.81011
Staging Study of Hepatocellular Carcinoma Based on Network Analysis and Random Forest Method
Xin Li
School of Mathematics and Physics, North China Electric Power University, Beijing
Received: Jan. 12th, 2019; accepted: Jan. 24th, 2019; published: Jan. 31st, 2019

ABSTRACT
Hepatocellular carcinoma (HCC) is an invasive malignant tumor. Although the diagnostic techniques and treatment levels of hepatocellular carcinoma have made great progress, the early diagnosis of HCC is still a huge challenge. In this paper, we attempt to analyze core genes associated with clinical staging by gene network for information on the discovery of early HCC patients and improving the diagnostic techniques and treatment levels of HCC. First, we selected the gene expression data of 219 patients with early postoperative HCC in the GEO database, performed differential expression analysis, and randomly divided the data into training set and test set. We use the genes of training set to clustering out five modules by weighted gene co-expression network (WGCNA), and performed functional enrichment and pathway enrichment analysis for each gene module. We found that the blue module is related to some biological processes such as cell proliferation, division, cycle and DNA replication initiation, replication, repair, and this module is also related to some pathways such as cell cycle, P53 signaling pathway, HTLV-I infection, hepatitis B. These processes and pathways are closely related to the occurrence and development of HCC. Therefore, we use the enriched genes of the module for PPI network analysis, and 10 core genes that we selected with high connectivity is BUB1B, CCNA2, CCNB1, CCNB2, CDC20, MAD2L1, MCM4, PCNA, RFC4, and TOP2A. Then through the supervised learning of core genes in random forests, a classification model of BCLC staging was established and then applied to the test set. The study found that the method has a great help for the classification of early patients, and the correct rate reached 95.52%, but for the patients in the middle and late stages. The classification effect is not very good. This study raises awareness of the pathogenesis and staging of HCC. And it provides a new direction for HCC targeted therapy.
Keywords:Hepatocellular Carcinoma, WGCNA, PPI Network, Random Forest
基于网络分析和随机森林方法的肝细胞癌分期研究
李鑫
华北电力大学,北京
收稿日期:2019年1月12日;录用日期:2019年1月24日;发布日期:2019年1月31日

摘 要
肝细胞癌(Hepatocellular Carcinoma, HCC)是一种侵袭性恶性肿瘤,尽管肝细胞癌诊断技术及治疗水平有了较大的进步,但对HCC的早期诊断依然是个巨大的挑战。在本文中,我们试图通过基因网络分析与临床分期相关的核心基因,用于对早期HCC患者的发现提供信息和提高HCC诊断技术及治疗水平。首先,我们选用GEO数据库中包含219例早期术后HCC患者的基因表达数据,进行差异表达分析,并且将数据随机分为训练集与测试集,其中训练集采用加权基因共表达网络(WGCNA)分析聚类出五个模块,对各基因模块进行功能富集和通路富集分析,我们发现其中blue模块与细胞增殖、分裂、周期以及DNA复制启动、复制、修复等生物过程相关,与细胞周期、P53信号通路、HTLV-I感染、乙型肝炎等通路相关,这些过程和通路均与HCC的发生发展密切相关。因此,选取模块的富集基因进行PPI网络分析,选取连通度较大的10个核心基因BUB1B、CCNA2、CCNB1、CCNB2、CDC20、MAD2L1、MCM4、PCNA、RFC4、TOP2A,通过随机森林对核心基因进行监督学习,建立BCLC分期的分类模型,然后应用于测试集,研究发现该方法对于BCLC早期患者的分类有很大程度的帮助,正确率达到95.52%,但是对于患者的中后期分类效果不是很理想。该研究提高了对HCC的发病机制和分期研究的认识,为HCC靶向治疗提供了新的方向。
关键词 :肝细胞癌,加权基因共表达网络分析,PPI网络,随机森林

Copyright © 2019 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 引言
肝细胞癌(HCC)是全球最常见的恶性肿瘤,占所有癌症病例的5%以上,是全球癌症死亡的第五大原因 [1] [2] 。在发展中国家最为普遍,其发病率呈上升趋势,具有术后转移和复发的比例较高,长期生存率较低的特点 [3] 。全世界几乎80%的HCC病例都存在由慢性肝炎、炎症和纤维化引起的肝硬化等致癌性损伤 [4] 。纤维化和肝硬化的其他病因因素如遗传性血色素沉着症或非酒精性脂肪肝疾病也对HCC的发展有潜在影响 [5] [6] 。由于在HCC早期阶段缺乏症状和疾病的快速进展,大约80%的HCC患者被诊断为晚期疾病 [7] 。一般来说,肝切除和原位肝移植被认为是HCC的唯一治疗方法,当肿瘤负荷无法通过手术切除时,HCC的预后较差 [8] 。尽管在过去几十年中做出了重大努力,但是干扰HCC进展的治疗选择非常有限,并且需要用于有效治疗的新型治疗策略。
由于近年基因组学、转录组学以及测序技术的蓬勃发展,加权基因共表达网络(Weighted Gene Co-expression Network Analysis, WGCNA)正逐渐在生物学研究领域拓展其应用面,目前该方法已成功应用于癌症相关研究。在前列腺癌中来应用该方法构建mRNA和microRNA表达网络 [9] ;该方法鉴定出ASPM基因为胶质母细胞瘤的新型分子生物标志物 [10] ,还用来构建了神经胶质瘤的促细胞分化和发芽信号相关的共表达网络 [11] 。分期系统一方面可以评估病人的预后从而选择正确的治疗方法,另一方面,它也是比较不同治疗试验的重要工具,恶性肿瘤的分期是选择和改善治疗方法的基础。好的分期系统须具备简单、应用方便、可重复性好,并且能够提供可靠的疾病自然病史的信息和根据不同的治疗组分类等特点。但是目前,各地不同的分期系统中,尚没有一个分期系统被一致认为是最完善的。本文借助基因表达数据,应用该方法与PPI网络结合筛选出核心基因,希望通过借助癌症发展的重要过程分析出与分期相关的特征基因,借助随机森林模型建立分类模型,深入学习癌症患者分期过程的分子学研究。
2. 材料与方法
2.1. 数据介绍
微阵列数据在Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo)上公开获得登录号GSE14520的肝癌数据。其中我们选取在96 HT HG-U133A 2.0微阵列平台上进行处理过的221例肿瘤样本以及220例正常样本。去掉缺失临床信息的患者后剩余219例肿瘤样本。临床信息包括性别(Gender)、年龄(Age)、HBV活动状态(HBV viral status)、丙氨酸转移酶(ALT)、肿瘤大小(Main Tumor Size)、结节型(Multinodular)、肝硬化(Cirrhosis)、切除前患者的血清AFP水平(AFP)和HCC预后分期系统巴塞罗那诊所肝癌(BCLC)、癌症肝意大利计划(CLIP)及肿瘤淋巴结转移(TNM)等。其中BCLC分期中有148例早期患者,22例中期患者,29例进展期患者及20例极早期患者。数据中探针总数是22,268个,将基因与探针对应,其中删除810个单个探针对照多个基因和1029个无基因对照的探针,整合单个基因对应多组探针的情况中,本文选取表达量最高的作为最终基因的表达量,最终得到12,742个基因。
2.2. 数据预处理
初始数据通常具有冗余性、不完整性和不规范性的特点,会影响到我们对数据的直接分析,如果数据中存在噪音干扰,还会造成结果的偏差。因此,本文对数据的处理采用分位数标准化 [12] 来去除掉芯片之间的系统误差。差异表达基因的选取过程采用R3.4.1中limma包 [13] 进行分析,选取|lFC|大于1,p值小于0.05的基因进行后续研究,筛选出927个差异表达基因,其中有332个上调基因,有595个下调基因。
将219个样本随机分为训练集和测试集,其中训练集含110个样本,测试集含有109个样本,由于因加权基因共表达网络的结果容易受到离群样本影响,所以训练集和测试集均采用芯片间相关度(inter-array correlation, IAC) [14] 方法来评估芯片数据的分布情况。训练集通过WGCNA方法 [15] 挖掘基因模块,将各个模块进行GO功能富集分析,了解各模块聚类原因,然后选取与HCC发展密切相关的模块,先取该模块GO功能富集的基因放入在线工具string中做PPI网络选取出连接度高的10核心基因。将这些核心基因建立随机森林模型,测试集用于测试集模型对于患者分期的分辨能力。
2.3. WGCNA方法
加权基因共表达网络分析(Weighted gene co-expression network analysis, WGCNA)是用于描述跨微阵列样品的基因之间的相关模式的系统生物学方法。网络中的每一个节点代表一个基因,如果在不同条件下基因之间的表达存在共性,那么这两个基因在同一个基因共表达网络,或在同一个模块(module)中。下面使用WGCNA [16] 方法构建HCC样本中基因的共表达网络。首先,计算基因共表达的相关矩阵:
(1)
其中 和 分别是基因和的基因表达量。通过两个量的皮尔森系数cor转化为相互作用矩阵 。再计算基因之间的邻接系数:
(2)
其中, 表示邻接系数,即通过 次方的幂指数运算对每对基因的相关系数进行加权。其中 称之为软阈值。这种转变旨在给予强联系更多的权重,并降低预测共表达网络中弱连接的重要性,以提高共表达网络的可靠性。最后,考虑到某个基因与分析中其他所有基因之间的关系,将邻接矩阵转换为拓扑矩阵 ,矩阵中的元素如下:
(3)
其中, 表示基因公共连接的节点之间邻接系数乘积的总和, 和 分别表示基因 、 与各自连接节点之间邻接系数的加和。通过节点间的相异程度 来衡量基因模块所具有的生物学意义,因此通过 来实现网络的构建。
2.4. 随机森林
随机森林(Random Forest),顾名思义就是由很多随机生成的树构成的森林,由于生成树是随机的,所以是相互独立的,彼此没有关联或者依赖性。随机森林分类的基本思想 [17] :第一步,利用Bootstrap抽样从原始训练集抽取k个样本,并且每个样本的样本容量均与原始训练集中相同;第二步,对k个样本分别建立k个决策树模型得到k种分类结果;第三步,依据k种分类结果对每个记录进行投票表决从而决定其最终分类。分类的正确率通过抽样过程中所形成的袋外数据(OOB)来进行预测,对每次的预测结果进行汇总便可得到错误率OOB估计,从而对组合分类的正确率进行评估。
随机森林中的每棵分类树都是二叉树,其生成遵循自上向下的递归分裂原则,即从根节点对训练集进行依次划分,在二叉树中根节点需包含所有的训练数据,并遵循节点不纯度最小原则分裂为左、右两节点,分别包含训练数据的一个子集,并且节点遵从相同规则继续分裂,直到满足分支停止规则停止生长。如果节点 上的分类数据均来自同一类别,则该节点的不纯度 。其中,不纯度度量方法为Gini准则 [18] ,即假定 为节点 上属于 类样本个数占训练样本总数的频率,则Girfi准则表示如下:
(4)
3. 统计分析
3.1. WGCNA分析
因加权基因共表达网络的结果对离群样本敏感,应去除其中的离群样本,因此本文采用了芯片间相关性(inter-array correlation, IAC) [14] 方法来评估芯片数据分布情况。其中,具有低平均IAC值或者无法在树形图上聚类的样本为离群样本。IAC方法:第一步,计算出每个芯片的平均芯片间相关性标记为A;第二步,计算出每个芯片平均芯片间相关性A的平均值标记为A';第三步,计算出所有芯片平均芯片间相关性A的标准差标记为sd;第四步,根据(A-A')/sd计算每个样本的偏倚度标记为num。
为保证网络构建结果的可靠性,首先需要对数据源进行质量控制,包括芯片表达数据预处理和异常样本的去除。借助IAC方法去除离群样本并根据样本聚类树高度衡量去除效果。我们经过三次上述步骤操作,见图1,样本的聚类树高度由高于0.6降至低于0.4。经处理后,训练集剩余99个样本,测试集剩余92个样本。
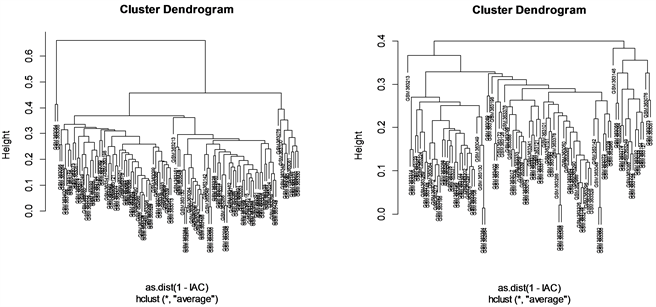
Figure 1. Training set tumor sample hierarchical clustering tree
图1. 训练集去除离群样本前后聚类树
加权基因共表达网络需满足无尺度网络条件,定义基因共表达矩阵中的元素是基因相关系数的加权值,选择权重标准是每个基因网络中包含基因之间的连接需服从无尺度网络分布(scale-free networks) [16] ,即连接数为i的概率p(i)与i的r次方成反比,即p(i)~i−r。研究选择合适的加权系数逼近无尺度网络分布,使得连接数i的节点对数值(log(i))与此节点出现概率的对数值(log(p(i)))呈负相关,相关系数至少应达到 0.8,在不同模块中基因的平均连接度较高才使得检测的模块更有意义。
接下来,我们选择适当的加权参数P,以便对邻接矩阵加权后使之符合无尺度网络标准。经过计算,在不同的软阈值情况下,绘制节点连接度的对数log(i)与该节点出现的概率的对数log(p(i))之间的相关系数图,图2中左图展示了不同软阈值对应的log(i)与log(p(i))之间的相关系数,系数越高表示网络越符合无尺度网络分布,右图则表示不同软阈值对应基因邻接系数的均值,反映了网络的平均连接水平。我们选择p = 5构建基因网络,log(i)与log(p(i))的相关系数接近0.8。我们分析绘制了网络中节点的连接度分布图以及log(p(i))与log(i)的散点图,从图3中可以看出线性回归结果符合无尺度网络标准,其相关系数为0.84。
我们经上述研究最终选取p = 5,然后按照WGCNA算法,计算HCC差异表达的相关矩阵、邻接矩阵、以及拓扑矩阵,然后经聚类分析得到基因的系统聚类树。根据动态混合剪切法 [19] ,第一步,同样设定单个基因模块最,小基因数为30,并选择中等程度(deep Split = 2)的分类方式构建初等网络;第二步,便求得每个基因模块的ME,并对ME进行聚类,再将相似度高的ME所对应的基因模块进行合并,确定五个基因模块。如图4所示五个模块分别为blue、brown、green、turquoise、yellow,并且模块中含有37到569个基因,23个基因未能分配到任何基因模块中。见表1,显示了各模块的基因数。我们采用模块内部连接度(Intramodular connectivity, IC)来描述特定的模块中的节点与模块中其他节点的关联程度,模块身份(modular membership, MM)来表示基因在相应模块中基因的重要性。图5中可以看出blue模块的IC与MM相关系数达到0.98。

Figure 2. Determination of soft thresholds in WGCNA
图2. WGCNA方法中软阈值的确定
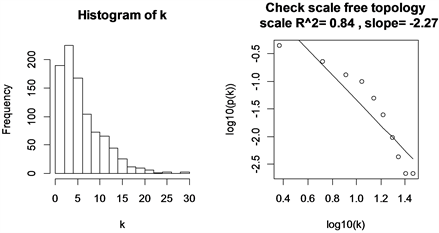
Figure 3. Scale-free network test with soft threshold power = 5 of training set
图3. 训练集软阈值为5时无尺度网络检验

Table 1. Training set clustering module and the number of genes in the corresponding module
表1. 训练集聚类模块及相应模块中的基因个数

Figure 4. Initial and final modules derived from dynamic shearing with the WGCNA method
图4. 训练集样本聚类树及初始划分、合并模块图
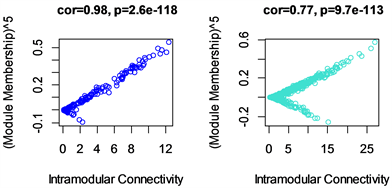
Figure 5. MM and IC relationship diagram between training set blue module and turquoise module
图5. 训练集blue模块和turquoise模块中MM与IC关系图
3.2. 基因模块的功能富集分析及通路分析
我们将每个模块的基因放入在线工具DAVID [20] (https://david.ncifcrf.gov/)进行GO功能和KEGG通路富集分析,以及相应的可视化分析。在五个模块富集结果中,我们发现blue模块中的基因参与了细胞周期、细胞分裂、细胞凋亡、以及p53信号通路等众多与癌症的发生发展的重要过程中。因此,为了深入了解HCC的分期发展,我们接下来对该模块的核心基因进行深入的研究分析。
首先,在GO术语富集过程中,blue模块基因分别富集到基因执行的分子功能(Molecular Function)、基因所处的细胞组分(Cellular Component)、基因参与的生物学过程(Biological Process)三大基因注释中,如图6所示。其中功能富集结果见表2显示,blue模块中的基因被富集到细胞分裂、有丝分裂核分裂、有丝分裂细胞周期的G1/S转换及G2/M转换、细胞增殖、姐妹染色单体凝聚力、DNA复制启动、DNA复制、DNA修复、对药物的反应、蛋白质SUMO化等重要的生物过程,如图7所示,左图展示的相应GO功能下基因的富集图,红点对应上调基因,蓝点对应下调基因,可以看出blue模块中的大部分基因为上调基因。右图为部分blue模块GO术语的ID号及对应的生物含义。
Blue模块基因参与到了细胞周期、卵母细胞减数分裂、p53信号通路、DNA复制、RNA转运、嘧啶代谢、错配修复、核苷酸切除修复等重要信号通路,见表3。

Figure 6. Three major functions of GO terminology for gene clustering in the blue module
图6. Blue模块中基因聚类的GO术语三大功能

Figure 7. Some GO function enrichment in the blue module and the gene up and down display in the corresponding process
图7. Blue模块中部分GO功能富集及对应过程中基因上、下调展示
Table 2. Some related processes of the blue module in GO function enrichment
表2. GO功能分析blue模块基因富集的相关生物学过程
Table 3. Some related paths of the blue module in the Kegg path enrichment process
表3. KEGG通路分析blue模块基因富集的相关通路
3.3. PPI网络分析和Cytoscape可视化分析
我们选取GO功能富集中且的生物过程的基因。通过在线工具STRING (https://string-db.org/)将得到的100个基因进行PPI网络 [21] 分析,通过Cytoscape工具 [22] 将网络可视化,选取连通度较大的前10个核心基因,进行深入学习。见表4,展示了所选的核心基因的相关连通度等信息。
Table 4. Inter-gene connectivity in the PPI network analyzed by the Cytoscape tool
表4. Cytoscape工具分析出的PPI网络中基因间的连通度
3.4. 随机森林对核心基因监督学习
我们通过网络分析得到重要模块基因的子集是相关的并且与癌症相关,但是构成该子集的基因对于癌症的重要性以及对于分期的作用是未知的,需要通过机器学习方法来发现和选择。随机森林分类算法本身可以对基因的重要性进行排序。我们将所选取的10个核心基因建立随机森林分类模型,通过对训练集的深度学习建立模型,然后通过测试集对模型进行检验。
我们分别绘制了基于OOB数据的模型误判率散点图以及相关误差与随机森林中决策树数量的关系图,如图8,在构造随机森林模型过程中,从散点图中可以看出,当mtry = 6时,其误判率较低,从而我们可以再进一步的确定应该使用的决策树数量。在右图中看出,当模型中决策树的数量小于400时,模型误差出现较大的波动,当决策树的数量大于400时,模型误差趋于稳定。所以我们可以将模型中的决策树数量大致确定为400左右来达到最优模型。我们还绘制出了随机森林模型中每棵树的节点个数柱形图,如图9所示,可以看出在构建的随机森林模型中,最小的决策树有12个节点,最大的决策树有26个节点,树之间的节点个数有所差异。
在随机森林分类问题中,randomForest包中提供了两个计算变量重要性的指标,一个为基于OOB,计算预测误差率的指标MeanDecreaseAccuracy,并且通过OOB数据进行验证,它在特征选择方面的可信度较高。另一个则是基于样本拟合的模型计算Gini系数的指标MeanDecreaseGini,在建立模型的过程中,通过计算每个变量在分叉节点不纯度的减少量之和来衡量变量重要性。从图10中可以看出基因中的细胞周期蛋白A2 (CCNA2)和细胞周期蛋白B1 (CCNB1)等变量较为重要,经研究发现这两个基因均参与到了免疫应答和细胞周期过程,并且在正常癌旁组织中和癌症中存在着不同的功能激活和抑制转换机制 [23] 。

Figure 8. Relationship between false positive rate scatter plot and related errors and the number of decision trees in random forests
图8. 误判率散点图和相关误差与随机森林中决策树数量的关系图

Figure 9. Column number of nodes
图9. 节点数柱状图

Figure 10. Random forest variable importance measure scatter plot
图10. 随机森林变量重要性测度散点图
Table 5. Classification results of training set and test set
表5. 训练集与测试集的分类结果
通过训练集建立模型,并对测试集进行预测,见表5,展示了预测结果同训练集和测试集实际结果之间的差别情况。列为数据真实的分期状况,行为预测的分期情况,可以看出训练集中处于A期真实有67例患者,预测正确的有64例,3例被预测为B期,预测的准确率高达95.52%;测试集处于A期的有67例患者,其中65例预测准确,2例被预测为B期患者,准确率高达97.01%。但是对于无分期患者以及中晚期的患者的分类效果不佳。最终得出随机森林分类率为70.65%。
4. 结论
本文主要研究了肝细胞癌的分期,旨在通过基因研究对HCC患者BCLC分期有进一步的了解,可以通过核心基因方向对于HCC患者的BCLC分期能够更为准确,从而对BCLC分期产生积极的推动作用。
本文建立了对BCLC分期的分类模型。首先借助WGCNA方法对差异表达基因进行聚类分析得出五个模块,经过对每个模块进行了富集分析后,发现blue模块中的基因参与了众多癌症发生发展过程。随后结合PPI网络,筛选出该模块中前十个核心基因,通过这是个随机变量建立随机森林分类模型,结果发现这是个基因对于BCLC分期早期患者的分类效果极佳,正确率达到95.52%,对于中晚期患者的分类效果不明显。
本文选出的十个模块的核心基因分别为CCNB1、TOP2A、RFC4、MAD2L1、PCNA、CCNB2、CCNA2、MCM4、CDC20、BUB1B。这些基因参与到细胞分裂、有丝分裂核分裂、有丝分裂细胞周期的G1/S转换及G2/M转换、细胞增殖、姐妹染色单体凝聚力、DNA复制启动、DNA复制、DNA修复、对药物的反应、蛋白质SUMO化等重要的生物过程以及细胞周期、卵母细胞减数分裂、p53信号通路、DNA复制、RNA转运、嘧啶代谢、错配修复、核苷酸切除修复等重要信号通路。我们将这些基因放入GO注释以及通路中,了解其对HCC的预后作用。我们的讨论如下:
1) 这些基因均参与到了GO:0000278注释有丝分裂细胞周期(mitotic cell cycle)的功能中,其中MCM4、PCNA、RFC4、TOP2A四个基因参与到了DNA复制(DNA replication)过程,细胞周期中,当DNA复制启动蛋白异常高表达,必然导致细胞快速完成的DNA复制,使得细胞进入高度增殖状态,例如:增生细胞核抗原(PCNA)基因的表达与诸多肿瘤的恶性程度、浸润、转移密切相关,PCNA蛋白的表达与肝脏肿瘤的恶性行为也存在密切联系 [24] ;研究表明,TOP2A蛋白表达水平与组织学分级、肿瘤大小、分子分型均具有相关性 [25] 。BUB1B、CCNA2、CCNB2三个基因参与到了有丝分裂(Mitosis)过程,癌症的形成过程中多种基因相互作用,恶性肿瘤均表现出增殖的活跃,所以HCC也不例外,因此使得涉及到细胞增生分裂的有丝分裂过程在肿瘤的生长进程中也格外重要。
2) 这些筛选的核心基因富集到了细胞周期、卵母细胞减数分裂、p53信号通路、DNA复制这些相关通路中。其中CCNB1和CCNB2均在p53信号通路中,这些蛋白质的调剂严重影响着细胞周期G2停滞(Cell cycle G2 arrest)。彭绍华等 [26] 研究表明细胞周期蛋白在肝细胞癌组织中呈不同程度的高表达,使得细胞周期缩短,癌细胞增生活跃,细胞凋亡减少,恶性表型增加。
致谢
在此向悉心指导我的导师张娟老师献上诚挚的谢意!在文章撰写的过程中,张老师给予我细心的指导和支持,培养了我解决问题的能力和克服困难的毅力,同时,老师严肃的科学态度和严谨的治学精神深深的感染着我,让我更加认真的对待科研和学习,也让我明白在未来的生活和工作中明白做任何事情都要认认真真,循序渐进!
文章引用
李 鑫. 基于网络分析和随机森林方法的肝细胞癌分期研究
Staging Study of Hepatocellular Carcinoma Based on Network Analysis and Random Forest Method[J]. 统计学与应用, 2019, 08(01): 95-107. https://doi.org/10.12677/SA.2019.81011
参考文献
- 1. El-Serag, H.B. and Rudolph, K.L. (2007) Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology, 132, 2557-2576. https://doi.org/10.1053/j.gastro.2007.04.061
- 2. Mikulits, W. (2018) Epithe-lial to Mesenchymal Transition in Hepatocellular Carcinoma. Future Oncology, 5, 1169.
- 3. 李保国. 肝细胞癌预后相关细胞分子生物标志物研究进展[J]. 国际肿瘤学杂志, 2015, 42(5): 395-398.
- 4. Kensler, T.W., Qian, G.S., Chen, J.G., et al. (2003) Translational Strategies for Cancer Prevention in Liver. Nature Reviews Cancer, 3, 321-329. https://doi.org/10.1038/nrc1076
- 5. Jou, J., Choi, S.S. and Diehl, A.M. (2008) Mechanisms of Disease Progres-sion in Nonalcoholic Fatty Liver Disease. Seminars in Liver Disease, 28, 370-379. https://doi.org/10.1055/s-0028-1091981
- 6. Wallace, D.F. and Subramaniam, V.N. (2009) Co-Factors in Liver Disease: The Role of HFE-Related Hereditary Hemochromatosis and Iron. Biochimica et Biophysica Acta (BBA)/General Subjects, 1790, 663-670. https://doi.org/10.1016/j.bbagen.2008.09.002
- 7. Sun, V. and Sarna, L. (2008) Symptom Management in Hepatocellular Carcinoma. Clinical Journal of Oncology Nursing, 12, 759-766. https://doi.org/10.1188/08.CJON.759-766
- 8. Tanaka, S. and Arii, S. (2010) Molecular Targeted Therapies in Hepatocellular Carcinoma. Hepatology, 48, 1312-1327.
- 9. Wang, L., Tang, H., Thayanithy, V., et al. (2009) Gene Networks and microRNAs Implicated in Aggressive Prostate Cancer. Cancer Research, 69, 9490-9497. https://doi.org/10.1158/0008-5472.CAN-09-2183
- 10. Horvath, S., Zhang, B., Carlson, M., et al. (2006) Analysis of Oncogenic Signaling Networks in Glioblastoma Identifies ASPM as a Molecular Target. Proceedings of the National Academy of Sciences of the United States of America, 103, 17402-17407. https://doi.org/10.1073/pnas.0608396103
- 11. Ivliev, A.E., ‘t Hoen, P.A.C. and Sergeeva, M.G. (2010) Coexpression Network Analysis Identifies Transcriptional Modules Related to Proastrocytic Differentiation and Sprouty Signaling in Glioma. Cancer Research, 70, 10060-10070. https://doi.org/10.1158/0008-5472.CAN-10-2465
- 12. Bolstad, B.M., Irizarry, R.A., Åstrand, M., et al. (2003) A Comparison of Normalization Methods for High Density Oligonucleotide Array Data Based on Variance and Bias. Bi-oinformatics, 19, 185-193. https://doi.org/10.1093/bioinformatics/19.2.185
- 13. Smyth, G.K. (2005) Limma: Linear Models for Microarray Data. Bioinformatics & Computational Biology Solutions Using R & Bioconductor, 397-420.
- 14. 王攀. 加权基因共表达网络分析(WGCNA)在食管鳞癌中的应用[D]: [博士学位论文]. 北京: 北京协和医学院中国医学科学院; 北京协和医学院; 中国医学科学院; 清华大学医学部, 2014.
- 15. Langfelder, P. and Horvath, S. (2008) WGCNA: An R package for Weighted Correlation Network Analysis. BMC Bioinformatics, 9, 559. https://doi.org/10.1186/1471-2105-9-559
- 16. 宋长新, 雷萍, 王婷. 基于WGCNA算法的基因共表达网络构建理论及其R软件实现[J]. 基因组学与应用生物学, 2013, 32(1): 135-141.
- 17. Kandaswamy, K.K., Chou, K.C., Martinetz, T., et al. (2011) AFP-Pred: A Random Forest Approach for Predicting Antifreeze Proteins from Se-quence-Derived Properties. Journal of Theoretical Biology, 270, 56-62. https://doi.org/10.1016/j.jtbi.2010.10.037
- 18. 武晓岩, 李康. 随机森林方法在基因表达数据分析中的应用及研究进展[J]. 中国卫生统计, 2009, 26(4): 437-440.
- 19. Langfelder, P., Zhang, B. and Horvath, S. (2008) Defining Clusters from a Hierarchical Cluster Tree: The Dynamic Tree Cut Package for R. Bioinformatics, 24, 719-720. https://doi.org/10.1093/bioinformatics/btm563
- 20. Marr, D. (1982) Vision: A Computational Investigation into the Human Representation and Processing of Visual Information. Quarterly Review of Biology, 8.
- 21. 李敏, 陈建二, 王建新. 基于复杂网络理论的PPI网络拓扑分析[J]. 计算机工程与应用, 2008, 44(8): 20-22.
- 22. Saito, R., Smoot, M.E., Ono, K., et al. (2012) A Travel Guide to Cytoscape Plugins. Nature Methods, 9, 1069-1076. https://doi.org/10.1038/nmeth.2212
- 23. 周慧蕾. CCNB1和CCNA2在人类正常邻近组织和肺癌中不同功能激活及抑制转换机制与网络构建[D]: [硕士学位论文]. 北京: 北京邮电大学, 2015.
- 24. 李立人, 施公胜, 孙超. PCNA和VEGF在肝细胞肝癌中的表达意义[J]. 世界华人消化杂志, 2005, 13(4): 560-561.
- 25. 华骁帆. 早期非特殊性浸润性乳腺癌TOP2a蛋白表达与分级、分期及分子分型相关性分析[D]: [硕士学位论文]. 苏州: 苏州大学, 2016.
- 26. 彭绍华, 杨剑锋, 谢平平, 等. 细胞周期蛋白在肝细胞癌组织中的表达及其与肿瘤细胞凋亡的关系[J]. 癌症, 2005, 24(6): 695-698.