Material Sciences
Vol.
09
No.
05
(
2019
), Article ID:
30288
,
11
pages
10.12677/MS.2019.95057
First-Principles to Study the Electronic Structure of CuAlO2 Materials
Hongjiang Li, Youzhun Fan, Dongxue Chen, Shuai Zhang, Tao Sun, Hao Pang, Zhi Liang, Xiang Liu
Faculty of Materials Science and Engineering, Kunming University of Science and Technology, Kunming Yunnan

Received: Apr. 28th, 2019; accepted: May 13th, 2019; published: May 20th, 2019

ABSTRACT
CuAlO2 materials with delafossite structure are one of the important p-type transparent conductive semiconductor materials. The crystal structure, energy band structure and state density, as well as optical and thermodynamic properties of CuAlO2 were investigated by First-principles method of Density Functional Theory. The effects of GGA(PBE) and LDA(CA-PZ) exchange-correlation functional algorithms in simulation calculation results are also compared. The indirect band gap value Egi = 1.638 eV and direct band gap value Egd = 3.635 eV of CuAlO2 were calculated by GGA (PBE). It is in good agreement with the experimental values. Furthermore, the absorption spectrum, reflection spectrum, free energy, enthalpy, entropy and specific heat capacity of CuAlO2 were obtained.
Keywords:First Principles, CuAlO2, Density Functional Theory, Energy Band, Density of State
CuAlO2电子结构的第一性原理研究
李洪江,樊有准,陈东学,张帅,孙涛,庞浩,梁志,刘翔
昆明理工大学材料科学与工程学院,云南 昆明
收稿日期:2019年4月28日;录用日期:2019年5月13日;发布日期:2019年5月20日

摘 要
铜铁矿结构CuAlO2材料是重要的p型透明导电半导体材料之一。基于密度泛函理论的第一性原理计算,对CuAlO2材料的晶体结构、能带结构和态密度以及光学性质和热力学性质进行研究,为CuAlO2材料的本征结构、内部机制和物理参量的相互作用规律提供理论基础。同时比较了GGA(PBE)和LDA(CA-PZ)两种交换关联泛函算法对模拟计算结果的影响。通过GGA(PBE)计算得到CuAlO2的间接带隙值Egi = 1.638 eV,直接带隙值Egd = 3.635 eV,与实验数值吻合较好。另外,还获得了CuAlO2的吸收谱和反射谱以及自由能、焓、熵、比热容等。
关键词 :第一性原理,CuAlO2,密度泛函,能带,态密度

Copyright © 2019 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 引言
透明导电材料因其特殊的性能,在平板显示、触摸屏、太阳能电池、智能窗等多领域具有广泛的应用,具有重要的社会经济价值和军事价值,现已投产的有In:O3:Sn、SnO2:F等。然而它们都是n型宽带隙半导体薄膜,可见p型材料的实际应用却受到限制。2015年刘宏燕等 [1] 阐述这是因为在所制备的p型氧化物薄膜中,均很难制造出拥有高迁移率的导电薄膜,因此在相应领域研发中限制了p型透明导电氧化物薄膜的实际应用。2009年何阿玲等 [2] 提出如果能制备出p型铜铁矿结构CuAlO2透明半导电薄膜材料,就开展一个透明氧化物半导体的新领域,制备出CuAlO2太阳电池材料、FET以及透明PN结等一系列器件。2018年7月26日小米8透明探索版发布,其中的一大突破,就是其背部透明的设计,让手机的内部构造一览无余,因此CuAlO2透明半导体材料可以做透明手机备选材料,将实现整个器件的透明性,促进研发新的光电子器件及相关领域的发展。
铜铁矿结构的CuAlO2膜具有p型透明导电特性,本文基于第一性原理计算(First-principles),以CuAlO2材料作为研究对象,探究与之相关的基本物理性能。而在CuAlO2的材料的研究工作中,我们使用全势线性缀加平面波(FLAPW)方法计算CuAlO2(3R)的能带结构。发现导带底位于Γ点而价带顶位于F点,价带顶和导带底不在k空间的同一点,表明CuAlO2属于间接带隙半导体材料。1997年日本的H. Kawazoe等 [3] 教授在实验得到的Egi和Egd值分别为1.8 eV和3.5 eV,Benk等教授测量的Egi为1.65 eV。2003年,GAO Shanmin等 [4] 教授在实验得到p型透明CuAlO2半导体薄膜,高导电性透明CuAlO2薄膜的成功制造表明,纳米技术将有助于提高p型透明半导体的导电性。2012年,YING Xu等 [5] 研究的CuAlO2能带结构显示出间接的基本带隙;获得了2.29 eV + U校正的间接带隙,接近于实验所得的间接带隙值2.99 eV。2014年,LIU Qijun等 [6] 根据F点的价带最大值(VBM)和Γ点的导带最小值(CBM)计算出CuAlO2的间接带隙为2.195 eV。2017年,LIU Weiwei等 [7] 计算GGA + U时,纯CuAlO2的间接带隙为2.721 eV。同年,LIU Ruijian等 [8] 人对于完美的CuAlO2晶体进行带隙计算,导带最小值和价带最大值不在互易空间的同一点,表明有间接带隙,这与之前的文献报道结果非常吻合。众所周知,使用GGA(PBE)方法计算时低于正常实验带隙值,Γ处的直接带隙为1.81 eV,小于~3.5 eV的实验值。而Katayama-Yoshida等使用全势能线性缀加平面波方法(FLAPW)计算CuAlO2的间接跃迁带隙宽度、直接跃迁带隙宽度以及缺陷能级,结果表明Al空位在CuAlO2中属于深能级缺陷,但是Cu空位是浅能级受主缺陷,所以认为Cu空位是p型铜铁矿结构CuAlO2透明半导电薄膜材料导电的主要因数之一。
本论文通过由BIOVIA (之前命名为Accelrys)研究和开发的Materials Studio (MS)计算材料软件,主要采用22个计算模块其中的CASTEP [9] (Cambridge Series Total Energy Package)模块功能,对CuAlO2材料进行第一性原理研究。对这种材料的晶体结构以及禁带宽度、电子态密度以及光学性质和热力学性质进行计算,对材料体系的内部机制和物理参量的相互作用提供理论解释,广义梯度近似GGA(PBE)泛函计算所得的Egi和Egd值与实验结果吻合较好。这对CuAlO2材料的理论研究和实验指导方面具有重要的意义。
2. 计算方法和模型构建
2.1. 计算方法
通过由BIOVIA研究和开发的MS计算材料软件,采用22个计算模块其中的CASTEP [9] 和Reflex模块功能计算和模拟完成的。采用总能量平面波赝势方法,将离子势用赝势替代,同时在晶体周期性势场中使用三维周期性边界条件,电子波函数通过平面波基组展开。密度泛函理论(Density functional theory, DFT)研究相关势和电子–电子相互作用的交换时我们采用局域密度近似(Local-density approximation, LDA)泛函和广义梯度近似(Generalized gradient approximation, GGA)泛函进行校正,并且其中GGA可以得到准确的稳定的基态能量和分子几何构型。目前这两种泛函是电子结构计算的比较准确的理论方法。
分别选用基于局域密度近似的LDA (CA-PZ)和基于广义梯度近似的GGA (PBE)交换关联近似法,几何优化和能量计算分别采用超软(Ultrasoft)赝势和模守恒(Norm-conserving)赝势。为了得到稳定精确的计算结果,首先对CuAlO2进行几何结构优化,得到能量最低状态,再在此基础上,对几何优化后的晶体进行能量计算,然后使用Reflex模块模拟出材料的XRD谱线。计算中选用Cu、Al、O的价电子组态分别为Cu:3d104s1,Al:3s23p1和O:2s22p4。采用BFGS算法对CuAlO2晶体结构进行结构优化,其结构优化收敛标准设置如下:1) 单原子能量的收敛标准为5.0 × l0−6 eV/atom;2) 原子间相互作用力的收敛标准设为0.01 eV/Å;3) 晶体内应力的收敛标准设为0.02 GPa;4) 原子最大位移收敛标准设为5.0 × 10−4 Å。设置的参数全部达到收敛标准,结构优化完成。为了保证体系总能量及晶体结构在平面波基水平的收敛,采用平面波截断能Ecut = 400.00 eV、36 × 36 × 36 FFT网格、8 × 8 × 8 Monkhorst-Pack grid 的K网格点设置;SCF标准设为1.0 × 10−6 eV/atom,自洽场采用Pulay密度混合法,自治场设为2.0 × l0−4 eV/atom;在对模型的能量计算中,采用模守恒赝势,平面波截断能Ecut = 650.00 eV,并选用40 × 40 × 40 FFT网格。5) 在对模型的能量计算中,采用模守恒(Norm-conserving)赝势,平面波截断能Ecut = 650.00 eV,并选用40 × 40 × 40 FFT网格;在自洽场运算中,采用了Pulay密度混合法,自治场设为2.0 × l0−4 eV/atom;系统总能量和电荷密度在Brillouin区的积分计算使用Monkhorst-Pack grid方案来选择K网格点为8 × 8 × 8。
2.2. 模型构建
本论文通过使用剑桥大学无机非金属材料数据库(ISCD FindIt)导入CuAlO2的R-3c晶体结构,如图1所示:CuAlO2晶格参数:a = b = 2.86 Å,c = 16.96 Å;a = b = 90˚,g = 120˚。以ab轴为平面的CuAlO2铜铁矿结构是以六角Cu层以及AlO2层平铺而成;c轴以Al为核心的AlO6八面体和哑铃O-Cu-O层堆垛排列,H. CkandPal等 [10] 提出存在3R和2H的多型结构。哑铃O-Cu-O层中Cu+的3d电子态与O2−的2p电子态之间杂化的增强,使CuAlO2结构的价带有效质量降低,价带更加分散,从而使其p型导电性得到增强。由于六角Cu层的高导电性质,导致CuAlO2铜铁矿结构的电导率具有各向异性,沿ab轴方向的电导率要高于c轴电导率。另一方面,相比于Cu2O结构,CuAlO2铜铁矿结构中Cu原子的配位数变小(即三维交联的Cu+变成了二维),所以带隙得到增加,因此材料在可见光范围内透明。CuAlO2铜层之间d10~d10弱相互作用,郭得峰等 [11] 研究表明这种相互作用在很大程度上决定了CuAlO2的光电性质。其能量优化的原胞晶格参数为a = b = c = 5.89 Å,a = b = g = 28.09˚,如图2所示。
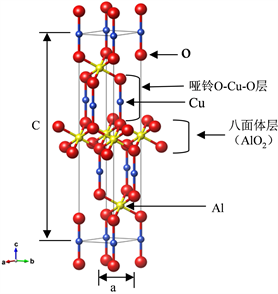
Figure 1. Crystal structure diagram of CuAlO2 (ISCD FindIt)
图1. CuAlO2晶体结构图(ISCD FindIt)
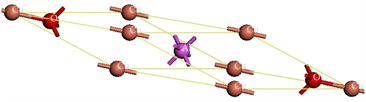
Figure 2. Optimization diagram of the primitive cell structure of CuAlO2
图2. CuAlO2优化原胞结构图
3. 结果与分析
3.1. XRD图谱与晶体结构
首先在Material Studio使用Reflex模块模拟出CuAlO2的X射线衍射谱,与实验测得的CuAlO2的X射线衍射谱相比较。如图3所示。从CuAlO2各个晶面衍射峰位、相对强度比较可知:理论计算模拟的CuAlO2的X射线衍射谱和实验测得的CuAlO2的X射线衍射谱两者吻合较好,而且可以通过标准铜铁矿结构进行索引,这可以为实验制备的CuAlO2材料提供物相检索依据。
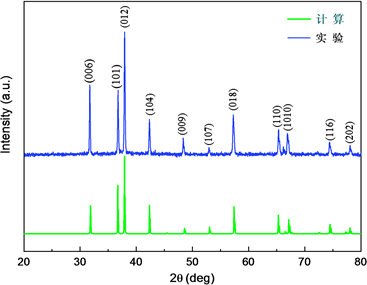
Figure 3. Experimental and theoretical simulation of the X-ray diffraction pattern of CuAlO2
图3. 实验及理论模拟CuAlO2的X射线衍射图谱
通过CASTEP模块对CuAlO2晶体结构进行结构优化,得到结构优化后的CuAlO2晶体的晶格常数如表1所示,同时表中一并给出了其他文献所报道的实验结果做对比。

Table 1. Comparison of simulation and experimental results of CuAlO2 lattice parameters
表1. CuAlO2晶格参数模拟和实验结果对比
从表1中可以看出,用GGA(PBE)近似方法和用LDA(CA-PZ)方法结构优化所得的优化晶格常数在标准值的基础上有所减小,两者相对标准值误差分别为:−0.578%和−0.723%;结构优化的晶格角度研究中,相对标准值的角度误差分别为:0.493%和−5.59%,可以看出GGA (PBE)计算的误差要比LDA(CA-PZ)小的多。可见,使用GGA(PBE)近似方法比使用LDA(CA-PZ)近似方法在晶体参数计算中更接近于实验值。而结构优化模拟后所得的CuAlO2的晶格常数与文献报道及实验值吻合较好,表明我们结构优化后的CuAlO2晶体结构是合理的,计算结果比较可靠。
3.2. 能带结构和态密度
为了进一步研究CuAlO2的电子结构属性,分别计算了CuAlO2的能带结构、总体态密度以及分波态密度。图4(a)、图4(b)、图4(c)和图4(d)分别是GGA (PBE)和LDA (CA-PZ)方法所得CuAlO2能带结构和总态密度图。由能带结构可知,CuAlO2是间接带隙半导体,GGA (PBE)方法计算得到F-G间的间接带隙值Egi = 1.638 eV,直接光学带隙值Egd =3.635 eV;LDA (CA-PZ)方法计算得到F-G间的间接带隙Egi = 1.564 eV,直接光学带隙Egd = 3.012 eV。而Benk等人测量的Egi为1.65 eV,以及H. Yanagi等 [12] 和H. Kawazoe等 [3] 得到的Egi和Egd值分别为1.8 eV和3.5 eV。本文GGA (PBE)方法计算所得值与以上实验得到的值非常接近,而LDA (CA-PZ)方法计算所得值与实验值相差较大,但ZHAO Yujun等 [13] 使用全势线性缀加平面波(FLAPW)方法计算CuAlO2间接跃迁带隙宽度是1.7 eV、直接跃迁带隙宽度为2.8 eV,可见用LDA计算的值与使用全势线性缀加平面波(FLAPW)方法计算得到的直接带隙接近。
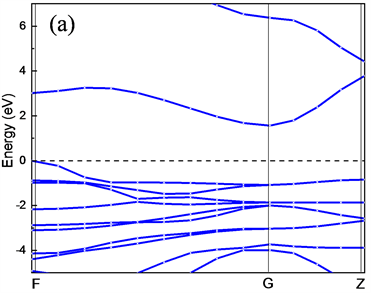
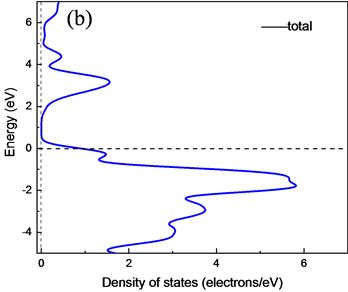
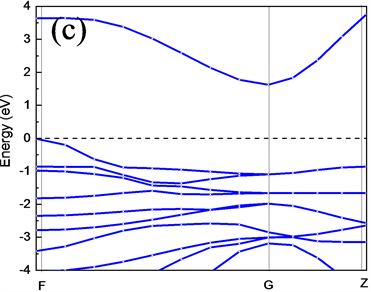
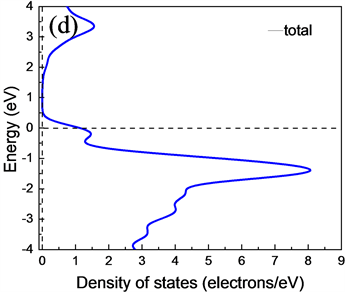
Figure 4. Energy band structure and total state density of CuAlO2 obtained by LDA and GGA methods (a), (b), (c) and (d) shown respectively
图4. (a)、(b)是LDA和(c)、(d)是GGA方法所得CuAlO2能带结构和总态密度图
表2为用GGA (PBE)和LDA (CA-PZ)计算出的CuAlO2的Egi和Egd与实验数据的比较和误差。可以看出使用GGA (PBE)方法计算所得的Egi和Egd相对误差分别为:−9.000%和3.857%,使用LDA(CA-PZ)方法计算所得的Egi和Egd相对误差分别为:−13.11%和13.94%。使用GGA(PBE)近似方法比使用LDA(CA-PZ)近似方法算出来的Egi和Egd与实验数值更接近。GGA(PBE)近似方法在计算CuAlO2电子结构方面要比LDA(CA-PZ)近似方法准确。
Table 2. Error of experimental and Egi and Egd data of calculated CuAlO2 by GGA and LDA
表2. 用GGA和LDA计算出的CuAlO2 Egi和Egd以及和实验数据的误差
图5(a)、图5(b)分别是分波态密度(PDOS)图和CuAlO2能带图,为便于分析,分别画出了Cu、Al和O元素各原子对应于能带结构的s、p和d轨道的态密度图。图6(a)、图6(b)和图6(c)分别是Cu、Al和O元素各轨道分波态密度图。计算中选用Cu、Al、O的价电子组态分别为Cu:3d104s1,Al:3s23p1和O:2s22p4。
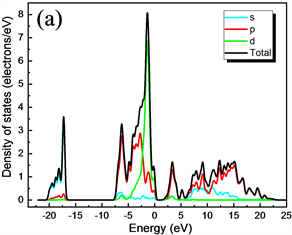
Figure 5. PDOS and CuAlO2 energy bands are shown by (a) and (b), respectively
图5. (a)、(b)分别是分波态密度(PDOS)和CuAlO2能带
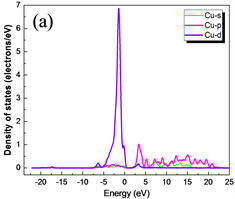
Figure 6. The wave density of each orbital of Cu, Al and O elements are shown by (a), (b) and (c), respectively
图6. (a)、(b)和(c)分别是Cu、Al和O元素各轨道分波态密度图
从能带图和各元素分波态密度图可以看出,CuAlO2的价带基本上可以分为两个区域:第一个是位于−20.14 eV~−17.18 eV的下价带,该部分主要是O的2s电子态密度的贡献,Al的3s和3p态也有少部分贡献;第二个是位于−7.33 eV~1.5 eV的上价带,这主要是Cu的3d态和O的2p电子态杂化的贡献,Al的3p态也有部分与其缠绕。导带部分,其主要分为两个区域:第一个是位于1.75 eV~5 eV,该部分主要是由Cu的3p轨道与O的2p杂化;第二个是位于5 eV之后,其主要是Al的3s及3p轨道缠绕,3s与3p电子态杂化的贡献。靠近能级的价带区域主要由Cu的3d轨道填充,靠近能级的导带区域主要由Cu的3p电子态杂化,可以看出在能级附近主要由Cu的p轨道与d轨道跃迁为主要影响。
3.3. 光学性质
当用光子能量足够大(紫外光、可见光、近红外光)的光照射样品时,可使其价带中电子跃迁到导带,形成电子–空穴对。其中,半导体材料的光吸收过程主要包括本征吸收(带隙吸收)、杂质吸收、激子吸收等。大量实验证明,本征吸收是半导体材料的主要吸收过程。
在线性响应范围内,固体宏观光学响应函数通常可由光的复介电函数
(1)
或复折射率
(2)
其中, ,
根据直接跃迁几率的定义和Kramers-Kroning色散关系我们可以推导处晶体介电函数虚部、实部、吸收系数、反射系数,具体结果如下:
(3)
(4)
(5)
(6)
其中C、V分别表示导带和价带,BZ表示第一布里渊区,K为倒格矢, 为普朗克常量, 为动量迁移矩阵元, 为角频率, 和 分别为导带和价带上的本征能级,n、k分别为折射率和消光系数。以上关系式分析晶体能带结构和光学性质的理论依据,它反映了能级间电子跃迁所产生光谱的发光机理。而且从某种意义上说,复介电响应函数 比宏观光学常数更能表征材料的物理特性,更易于与物理过程的微观模型及固体的微观电子结构联系起来。
本文计算中,CASTEP提供了三种可选项:1) 极化情况,即可以指定垂直入射光的极化方向;2) 非极化情况,即垂直入射光不存在极化方向,是各种极化光的平均值;3) 多晶情况,即各种方向入射光的平均,我们此次计算采用了第二种情况。入射方向选(100)方向,得到CuAlO2的反射谱、吸收谱。如图7分别以波长为横坐标计算所得到的CuAlO2的反射谱和吸收谱,图8分别以能量为横坐标计算所得到的CuAlO2的反射谱和吸收谱。它们分别反映了材料对不同波长(能量)光反射率和材料对不同波长(能量)光吸收情况。
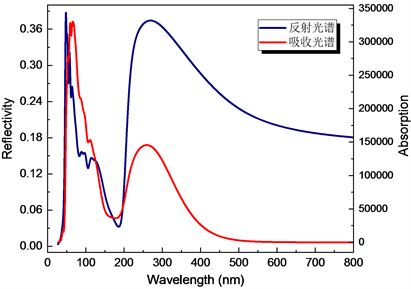
Figure 7. The absorption spectrum and reflection spectrum of CuAlO2, respectively
图7. 分别为CuAlO2的反射光谱和吸收光谱
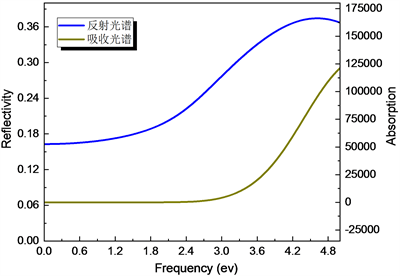
Figure 8. The reflection spectrum and absorption spectrum of CuAlO2, respectively
图8. 分别为CuAlO2吸收光谱和反射光谱
由 可知可见光的光子能量范围大约为1.5 eV~3.5 eV之间,从反射光谱和吸收谱可知,在400 nm~800 nm的可见光频段,材料的反射率较小且对光基本没有吸收,由此可以推测在400 nm~800 nm的可见光频段CuAlO2材料对可见光的透过率比较高,这也是为何CuAlO2是透明材料的原因。从定性分析来看,这与实验值吻合得很好。CuAlO2吸收谱还表明:在紫外光和微波波段,CuAlO2对其的吸收强度很大,透波性能较差。因此,CuAlO2在紫外光波段不透明或半透明。
3.4. 热力学性质
根据量子力学声子散射理论,总比热容可表达为:
(7)
其中Cqs是qs模式声子的比热容,h为约化的普朗克常数,wqs为第s支格波位于波矢q处的频率, 是波色–爱因斯坦分布函数,kB是波尔兹曼常数,T是绝对温度。由上式,可以得出摩尔比热容与温度的关系。
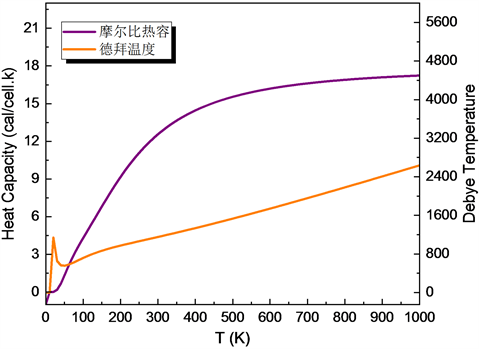
Figure 9. The Kelvin temperature dependence of molar specific heat capacity of CuAlO2 and the Debye temperature dependence of temperature of CuAlO2, respectively
图9. 分别为CuAlO2的摩尔比热容与开尔文温度的关系和德拜温度与温度的的关系
从图9可以看出,比热容随温度的变化在低温时是呈三次方关系即德拜的T3律,高温时趋于常数19.5 cal/(cell.K)即杜隆–珀蒂定律。
在理解材料热力学性质时,通常用到德拜温度的概念。在不同温度,用实际得出的真实比热容与德拜模型预测的比热容相比,从而得到德拜温度与温度的关系。德拜预测比热容具体为:
(8)
式中的 表示由德拜模型预测的等体积比热容, 是德拜温度。在不同的温度处,令由比热容(C)等于 ,从而得到不同温度处的德拜温度。
从图9可知,在20 K时,出现了德拜温度的极小值约为280 K。20 K以后,德拜温度基本随温度T呈线性增加趋势。温度从25℃到1000℃,德拜温度约从830.5 K递增到2044 K。另外,计算得到的CuAlO2自由能、焓和熵的变化如图10所示。
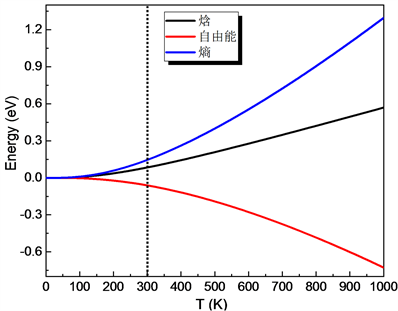
Figure 10. The change of free energy, enthalpy and entropy of CuAlO2
图10. CuAlO2自由能、焓和熵的变化
铜铁矿结构的CuAlO2具有p型透明导电特性,本文通过GGA(PBE)计算得到CuAlO2的间接带隙值Egi = 1.638 eV,直接带隙值Egd = 3.635 eV,推测在400 nm~800 nm的可见光频段CuAlO2材料对可见光的透过率比较高,以及CuAlO2自由能、焓和熵的变化等等。基于这些物理特性广泛用于太阳能电池、平面显示、电磁防护罩、功能窗、传感器等,在航空航天、军事武器等其它光电器件领域研发上也是值得参考的材料。
4. 结论
基于密度泛函理论(DFT)的第一性原理计算,研究了CuAlO2材料的晶格参数、电子结构、能带结构、态密度以及光学性质等基本物理性质。采用GGA(PBE)与LDA(CA-PZ)两种近似方法,对CuAlO2晶体结构进行结构优化,得到了CuAlO2的晶格常数a = b = 5.85 Å和c = 5.85 Å,α = β = 28.23˚和γ = 26.52˚,与实验值比较,晶格常数a的相对误差为−0.578%、c为−0.723%,角度α相对误差为0.493%、γ为−5.59%。而在GGA(PBE)与LDA(CA-PZ)两种算法的比较方面,在对于CuAlO2的结构优化模拟中,GGA(PBE)方法要比LDA(CA-PZ)算出的结果更接近实验数值。电子结构计算得出CuAlO2是间接带隙半导体,GGA(PBE)方法计算得到Egi = 1.638 eV,Egd = 3.635 eV;LDA(CA-PZ)方法计算得Egi = 1.564 eV,Egd = 3.012 eV。GGA(PBE)方法计算所得的Egi和Egd值与实验数值吻合较好。计算获得CuAlO2各个不同轨道在价带与导带上的具体分布;价带分为上价带和下价带,下价带主要是O的2s电子态密度的贡献;上价带主要是Cu的3d态和O的2p电子态杂化的贡献。导带部分主要是Cu的3p态与O的2p态以及Al的3s和3p电子态杂化的贡献。
最后,讨论了CuAlO2的光学性质和热力学性质,得到了CuAlO2的吸收谱和反射谱,从吸收谱和反射谱解释了CuAlO2是透明半导体的原因。另外,本文还得到了CuAlO2材料自由能、焓、熵、比热容和德拜温度随温度T在0 K~1000 K的变化曲线。
致谢
感谢刘翔老师的悉心指导。刘老师是我在模拟计算道路上的引路人,通过刘老师的耐心引领和积极辅导,我深深爱上了材料领域的模拟计算。其次,感谢师兄对我在MS软件上遇到的疑惑和不解,给予耐心热情的解释和帮助,大大增强了我对CASTEP的认识,提高了我对Materials studio软件的使用技能。
基金项目
云南省大学生创新创业训练计划项目资助(201710674204)。
文章引用
李洪江,樊有准,陈东学,张 帅,孙 涛,庞 浩,梁 志,刘 翔. CuAlO2电子结构的第一性原理研究
First-Principles to Study the Electronic Structure of CuAlO2 Materials[J]. 材料科学, 2019, 09(05): 447-457. https://doi.org/10.12677/MS.2019.95057
参考文献
- 1. 刘宏燕, 颜悦, 望咏林, 等. 透明导电氧化物薄膜材料研究进展[J]. 航空材料学报, 2015, 35(4): 63-82.
- 2. 何阿玲, 王新强, 刘高斌, 等. Mg掺杂CuAlO2电子结构的第一性原理研究[J]. 原子与分子物理学报, 2010, 27(1): 135-140.
- 3. Kawazoe, H., Yasukawa, M., Hyodo, H., et al. (1997) P-Type Electrical Conduction in Transparent Thin Films of CuAlO2. Nature, 389, 939-942. https://doi.org/10.1038/40087
- 4. Gao, S.M., Zhao, Y., Gou, P.P., Chen, N. and Xie, Y. (2003) Preparation of CuAlO2 Nanocrystalline Transparent Thin Films with High Conductivity. Nanoteehnology, 14, 538-541. https://doi.org/10.1088/0957-4484/14/5/310
- 5. Ying, X., Zhi, M.N. and Yuan, D.W. (2012) First-Principles Study of Nitrogen-Doped CuAlO2. Physics Letters A, 376, 2613-2616. https://doi.org/10.1016/j.physleta.2012.07.009
- 6. Liu, Q.J., Liu, Z.T. (2014) Theoretical Insights into Structur-al-Electronic Relationships and Relative Stability of the Cu-Al and O-Terminated CuAlO2 (0001) Surfaces. Vacuum, 107, 90-98. https://doi.org/10.1016/j.vacuum.2014.04.009
- 7. Liu, W.W., Chen, H.X., Liu, C.-L. AND Wang, R. (2017) Influence of Nitrogen-Doping Concentration on the Electronic Structure of CuAlO2 by First-Principles Studies. Physics Letters A, 381, 520-523. https://doi.org/10.1016/j.physleta.2016.11.032
- 8. Liu, R.J., Li, Y.F., Yao, B., et al. (2017) Shallow Acceptor State in Mg-Doped CuAlO2 and Its Effect on Electrical and Optical Properties: An Experimental and First-Principles Study. American Chemical Society, 9, 12608-12616. https://doi.org/10.1021/acsami.7b01354
- 9. Clark, S.J., Segall, M.D., Pickard, C.J., et al. (2005) First Principles Methods Using Castep. Z Kristallogr, 220, 567-570. https://doi.org/10.1524/zkri.220.5.567.65075
- 10. Kand Pal, H.C. and Seshadri, R. (2002) First-Principles Eleetronie Strueture of the Delafossites ABO2 (A=Cu, Ag, Au; B=Al, Ca, Sc, In, Y): Evolution of d10-d10 Interactions. Solid State Sciences, 4, 1045-1052. https://doi.org/10.1016/S1293-2558(02)01363-8
- 11. 郭得峰, 耿伟刚, 兰违, 等. Y掺杂A12O3高K栅介质薄膜的制备及性能研究[J]. 物理学报, 2005, 54(12): 5901-5906.
- 12. Yanagi, H., Moue, S.I. and Hamada, N. (2000) Electronics Structure and Optoelectronic Properties of Transparent P-Type Conducting CuAlO2. Journal of Applied Physics, 88, 4159. https://doi.org/10.1063/1.1308103
- 13. Zhao, Y.J. and Zunge, A. (2004) Electronic Structure and Ferromagnetism of Mn-Substituted CuAlS2 CuGaS2 CuInS2 CuGaSe2 and CuGaTe2. Physics Review B, 69, 104422. https://doi.org/10.1103/PhysRevB.69.104422