Journal of Organic Chemistry Research
Vol.
09
No.
02
(
2021
), Article ID:
43177
,
9
pages
10.12677/JOCR.2021.92003
Akt激酶小分子抑制剂研究进展
——ATP竞争性抑制剂研究概述
王彦凯,黎文海
中国药科大学理学院,江苏 南京

收稿日期:2021年4月14日;录用日期:2021年6月10日;发布日期:2021年6月17日

摘要
PI3K-Akt-mTOR信号通路是调节细胞周期活动的重要通路之一,该通路的失调是导致肿瘤产生的原因之一。目前以Akt为靶点的小分子抑制剂已经成为抗肿瘤药物研发的思路之一,在近二十年中,研究人员通过高通量筛选及计算机辅助药物设计等方法,设计、优化并合成了大量有选择性和高活性的Akt小分子抑制剂,本文对近年来文献所报道的ATP竞争性抑制剂按照其结构类型进行综述。
关键词
PI3K/Akt信号通路,ATP竞争性抑制剂,抗肿瘤药物
Research Progress of Small Molecule Inhibitors of Akt Kinase
—Overview of ATP Competitive Inhibitor Research
Yankai Wang, Wenhai Li
School of Science, China Pharmaceutical University, Nanjing Jiangsu

Received: Apr. 14th, 2021; accepted: Jun. 10th, 2021; published: Jun. 17th, 2021

ABSTRACT
The PI3K-Akt-mTOR signaling pathway is one of the important pathways that regulate cell cycle activities, and the imbalance of this pathway is one of the causes of tumors. Small molecule inhibitors targeting Akt have become one of the ideas for the development of anti-tumor drugs. In the past two decades, researchers have designed, optimized and synthesized a large number of selective and highly active Akt small molecule inhibitors through high-throughput screening and computer-aided drug design. This article reviews the ATP-competitive inhibitors reported in the literature in recent years according to their structure types.
Keywords:PI3K/Akt Signaling Pathway, ATP Competitive Inhibitor, Antitumor Drugs

Copyright © 2021 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/


1. 引言
Akt是一种丝氨酸/苏氨酸激酶(也被称为PKB或蛋白激酶B)。Akt在细胞内进行信号传导,对促进细胞存活和增殖起着重要作用 [1]。Akt是磷脂酰肌醇-3-激酶(PI3K)信号传导途径的关键下游成分 [2]。研究表明,当胞外信号与细胞膜上的受体(通常是RTK,酪氨酸激酶受体)结合后,通过磷酸化衔接蛋白IRS1来激活PI3K (磷脂酰肌醇-3激酶),进而催化胞质内的PIP2 (磷脂酰肌醇-4,5-二磷酸)转化生成PIP3 (磷脂酰肌醇-3,4,5-三磷酸),PIP3作为第二信使将信号通过特异性识别PH结构域与Akt结合,通过招募PDK1并且介导mTOR2 (雷帕霉素靶蛋白复合物)的磷酸化来完全激活Akt [3]。研究表明,通过PI3K-PKB-mTOR细胞内激酶的信号传导是控制细胞增殖和存活的主要组成部分,而该途径的失调与几种人类癌症的发展密切相关 [4]。
2. ATP竞争性Akt小分子激酶抑制剂分类
近些年以Akt为靶点的小分子抑制剂作为药物化学研究的一个热点,相关科研工作者研发并且报道了许多相关Akt抑制剂的设计、合成及后续的活性检测,根据其与激酶的作用位点和抑制方式可以将Akt抑制剂大致分为ATP-竞争性抑制剂、变构抑制剂、磷脂酰肌醇类似物抑制剂、假底物抑制剂和其他类型抑制剂。其中ATP-竞争性抑制剂作为Akt小分子激酶抑制剂研究领域的热点之一,近年来众多该类型抑制剂被研发报道,ATP竞争性抑制剂是通过直接作用于激活的Akt的ATP结合位点,与ATP竞争性结合Akt,从而阻碍活化的Akt对下游底物的磷酸化。本文将对近年来被报道的ATP-竞争性抑制剂按其结构类型进行分类总结。
2.1. 氨基恶二唑类
GSK690693是葛兰素史克公司自主研发的一种ATP竞争性抑制剂。通过先导化合物1和2进行先导优化研究,并从中找到一个对Akt三种亚型都有很好抑制效果的化合物3。GSK690693是一种新型的ATP竞争性pan-Akt激酶抑制剂,其对Akt1、Akt2和Akt3的半数抑制浓度(IC50)分别为2 nM、13 nM和9 nM。研究还表明GSK690693在肿瘤细胞的亚群中抑制增殖并诱导凋亡,其活性与细胞内对Akt激酶活性的抑制相一致。在体内也表现出药效活性,并已显示出可抑制BT474肿瘤异种移植物的生长。 [5] [6] GSK690693是第一个进入临床试验的氨基恶二唑类ATP竞争性抑制剂,主要应对血液瘤和实体瘤的治疗。但后续的研究表明GSK690693在体内可能存在抑制糖原合成或激活糖原分解的作用从而引起高血糖产生,因此不得不终止临床I期试验 [7] (见图1)。
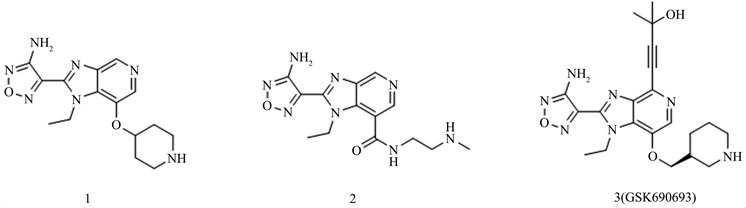
Figure 1. The structure of amino oxadiazole compounds (compounds 1~3)
图1. 氨基恶二唑类化合物结构(化合物1~3)
2.2. 吡咯并嘧啶类
辉瑞公司通过高通量筛选发现了以吡咯并嘧啶为母核的化合物4对Akt1的酶抑制活性达到290 nM,并以该化合物作为母核对其进行了结构修饰和相关构效关系的探究,通过其与Akt1激酶结构域结合生成的X射线共晶体结构显示吡咯并嘧啶实际上是与Akt1的铰链区结合的氢键供体–受体。晶体结构进一步揭示了三唑与结合水形成的分子间氢键可能是使其活性减弱的重要原因,因此将氢键替换为共价键从而将刚性引入分子中产生新的环,并得到了相较于化合物4抑制率提高7倍的化合物5,分析发现化合物5在吡咯并嘧啶C5处的氯取代发挥了重要作用,代表了拮抗Akt1的关键效价 [8]。
随后通过对化合物5的X射线共晶结构分析,发现含有螺二吲哚啉结构的系列化合物的抑制活性表现突出,含有氰基的化合物6的IC50为2.4 ± 0.6 nM,细胞药效为50 ± 19 nM。虽然化合物6对肿瘤生长具有良好的抑制效果,但其安全性和激酶选择性都较差,基于此Kevin D. Freeman-Cook等人利用“high speed parallel chemistry”展开了新一轮的结构探索,并从数千种结构中发现了3-氨基吡咯烷作为环胺部分在该系列化合物中显示出的独特选择性,但其抑制效率很差,因此通过进一步设计得到化合物7,其对Akt的选择性大大提高,而且对Akt1的IC50值为0.5 nM,是一种广泛选择性的,有效的,ATP竞争性Akt抑制剂,目前正处于临床试验中 [9] (见图2)。

Figure 2. The structure of pyrrolopyrimidine compounds (compounds 4~7)
图2. 吡咯并嘧啶类化合物结构(化合物4~7)
CCT128930 (化合物8)是基于药物片段筛选而得到的高选择性(PKA/Akt = 28)和高抑制效力(IC50 = 6 nM)的抑制剂 [10]。CCT128930虽然在细胞测定中具有活性,但在体内进行代谢时会导致快速清除和较低的生物口服利用度,因此对CCT128930进行了进一步的结构优化修饰得到化合物9。化合物9是有效的和口服可生物利用的PKB抑制剂,对Akt的IC50值为2.2 nM。其以良好的耐受剂量调节了体内PKB信号转导的生物标志物,并强烈抑制了裸鼠中人肿瘤异种移植物的生长 [11]。研究表明,化合物9是所有三种Akt亚型的有效酶抑制剂,并且是人MDAMB468细胞中Akt糖原合酶激酶3β (GSK3β)磷酸化的中度有效抑制剂。但基于酶活性其对于ROCK2的酶选择性不足,且对于hERG离子通道的活性的IC50为5 μM,易导致心源性猝死。因此Matt Addie等人在化合物9的基础上发现了临床候选药AZD5363 (化合物10),该化合物显示出增强的功效,降低的hERG亲和力和对密切相关的AGC激酶ROCK的更高选择性。该化合物表现出良好的临床前药物代谢和药代动力学(DMPK)特性,口服给药后,体内Akt和下游生物标记的磷酸化具有药效学抑制作用,并在乳腺癌异种移植模型中抑制了肿瘤的生长 [12] (见图3)。
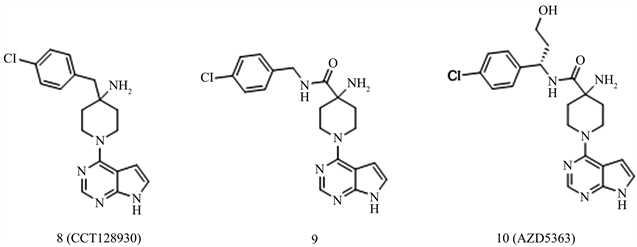
Figure 3. The structure of pyrrolopyrimidine compounds (compounds 8~10)
图3. 吡咯并嘧啶类化合物结构(化合物8~10)
2.3. 吡唑并嘧啶类
具有选择性的p70S6K (化合物11)是一类吡唑并嘧啶类Akt抑制剂,并发现当用溴取代吡唑并嘧啶上的3位乙基时可能会导致其选择性降低和酶促Akt活性更高。将其与已知Akt抑制剂进行叠合分析发现,在氯取代基所在的位置上引入一个疏水基团进入该口袋,从而有可能较大地增强Akt激酶的效力,基于此设计筛选出化合物12和化合物13。化合物12和13是能够抑制p70S6K和Akt1的双靶点抑制化合物,对Akt1的IC50分别达到5 nM和1 nM,并且能够显著抑制前列腺癌细胞中的GSK3β磷酸化。同时在不同的生物体中也都具有良好的药代动力学特征,口服利用度分别达到了58%和77%。由于化合物13在体外和体内的优异活性以及在较高物种中良好的口服生物利用度,因此被提名为临床候选药物,并以XL418的名称作为用于实体瘤和血液系统恶性肿瘤患者的口服制剂而得到开发 [13] (见图4)。

Figure 4. The structure of pyrazolopyrimidine compounds (compounds 11~13)
图4. 吡唑并嘧啶类化合物结构(化合物11~13)
2.4. 氨基嘧啶类
以氨基嘧啶为母核,设计并筛选出活性最好的化合物14。化合物14对多种激酶的中等至出色的选择性,但对AGC家族激酶的选择性很差 [14]。因此基于原先化合物设计并合成了一系列新的2-取代噻唑羧酰胺作为针对Akt的所有三种同工型的有效泛抑制剂,并得到了抑制活性较好的化合物15,其对Akt1,Akt2和Akt3的IC50值分别达到33 nM,1.95 μM和83 nM。该化合物还有效抑制下游MDM2和GSK3b蛋白的磷酸化,并在前列腺癌细胞中显示出强大的抗增殖活性 [15] (见图5)。

Figure 5. The structure of aminopyrimidine compounds (compounds 14~15)
图5. 氨基嘧啶类化合物结构(化合物14~15)
基于分子对接的QSAR模型来对Akt抑制剂分子进行结构优化。以4-氨基嘧啶为母核得到化合物16,其对Akt1的IC50达到7.7 nM,并对肿瘤细胞HCT116和OVCAR-8具有良好的抗增殖活性 [16] (见图6)。

Figure 6. Structure of Aminopyrimidine Compounds (Compound 16)
图6. 氨基嘧啶类化合物结构(化合物16)
2.5. 吡唑类
基于片段筛选和计算机辅助药物设计,发现了一种新型的、低分子量的能够有效抑制Akt活性的先导化合物17,其IC50值达到80 μM。以1H-吡唑作为该系列化合物母核,对配体有效类似物快速修饰,得到抑制活性较好的化合物18,其IC50值达到18 nM,相较于最初的先导化合物17抑制活性得到了大幅增强 [17]。运用高通量X射线晶体学和基于药物片段的分子对接技术设计并合成了化合物19 (AT13148)。AT13148是一种口服的ATP竞争性多AGC激酶抑制剂,对Akt1的IC50为38 nM。这种口服生物利用药物候选物实现了活性肿瘤暴露,在体内和体外具有临床相关遗传缺陷的癌细胞中诱导了强大的药效生物标志物调节和凋亡,并在多种人类肿瘤异种移植模型中展现出抗肿瘤功效。目前,AT13148正处于I期临床试验中 [18]。
GSK2110183 (化合物20)和GSK2141795 (化合物21)是一类新型的口服可用,具有ATP竞争性的泛Akt激酶抑制剂。其中GSK2110183对Akt1、Akt2和Akt3的IC50值分别为0.08 nM、2 nM和2.6 nM,GSK2141795对Akt1、Akt2和Akt3的IC50值分别为0.066 nM、1.4 nM和1.5 nM。两种化合物均在体外和体内抑制Akt途径和各种肿瘤细胞的增殖,此外,它们与先前报道的具有ATP竞争能力的Akt激酶抑制剂相比具有更有利的葡萄糖动态平衡特性。研究还发现,当Akt抑制剂与MEK抑制剂组合,与任一单一药物相比,GSK1120212均提高了疗效,表明了这类Akt抑制剂既可以单独用于临床,也可以与MEK抑制剂联用,为癌症治疗用药提供了新思路。目前,GSK2110183和GSK2141795处于I期和Ⅱ期的临床试验中 [19] [20]。
2016年,浙江大学的Wenhu Zhan等研究人员以GSK2141795 (化合物20)为先导化合物,设计、合成了一系列新颖的吡唑–呋喃羧酰胺类似物,并对它们的Akt1抑制活性以及抗增殖功效进行了生物学评估。针对HCT116和OVCAR-8细胞系,大多数化合物显示出中等至出色的Akt1抑制活性,以及良好的细胞毒性。其中化合物22抑制活性最好,其对Akt1的IC50值为61 nM,同时发现其对Akt2,Akt3,ROCK1和PKA都有一定抑制作用。此外,蛋白质印迹分析则表明化合物23可以显著抑制PC-3细胞中Akt底物GSK3β和LNCaP细胞中PRAS40的磷酸化水平 [21]。
2019年,上述课题组同样以GSK2141795 (化合物20)和化合物23为先导化合物,通过构象限制策略和分子对接技术,设计并获得了一系列的3,4-二取代哌啶衍生物。该系列化合物在体外和体内均显示出强大的抗肿瘤作用,但另一方面表现出不利的安全性特征,如低MTD,高hERG抑制和胃肠道出血。因此,对该系列化合物进行了构效关系的探究,并从中找到了具有对Akt1显著抑制的化合物24。化合物24显示出增强的Akt1效力和癌细胞抑制作用,显着降低了人类与人为相关的基因阻滞作用,并显着改善了安全性。同时还表现出良好的激酶选择性和良好的药代动力学特征,并且显示出非常有效的体内抗肿瘤功效,在SKOV3异种移植模型中具有超过90%的肿瘤生长抑制作用。在进行的进一步的机理研究中,证明了化合物24可以显着抑制异种移植模型中细胞和肿瘤组织中Akt激酶下游蛋白的磷酸化 [22] (见图7)。

Figure 7. The structure of pyrazole compounds (compounds 17~24)
图7. 吡唑类化合物结构(化合物17~24)
2.6. 哌嗪类
通过高通量筛选和基于结构的药物设计发现了哌嗪类先导化合物25,其对Akt1的IC50达到2.1 μM。研究发现在该结构中存在一对顺反异构体,对其拆分后发现相较于S型化合物,R型的化合物对Akt1的抑制活性增强了10倍左右,有着显著的提高,说明在该系列结构中,构型的改变对Akt活性抑制起着关键的作用。随后,通过Akt2的X射线结构,将先导化合物与靶点蛋白进行分子对接,得到化合物26,该结构对Akt三种亚型都有一定抑制作用,其IC50值分别达到20 nM,118 nM和179 nM。接下来以化合物26为先导化合物,对铰链区进行了优化设计分析,得到化合物27,其对Akt1的IC50值达到5 nM。在确定铰链区后,对化合物27进行三个关键部位地进一步构效关系探究,得到化合物28和化合物29,其对Akt1的IC50值都达到了1 nM,且对Akt的三个亚型都表现出了强大的酶和细胞抑制作用,同时在肿瘤异种移植物中显示出出色的p-PRAS40敲除能力,并具有高溶解度和良好的ADME性能。但不足之处是对其他相关激酶的选择性较差,特别是PKA、ROCK1和ROCK2的选择性缺乏使其耐受性差,不适合用作治疗剂,因此还有待进一步研究 [23] (见图8)。

Figure 8. The structure of piperazine compounds (compounds 25~29)
图8. 哌嗪类化合物结构(化合物25~29)
在化合物28的基础上,通过使用先前的SAR和结合位点的氨基酸序列分析,对铰链区进行改造,设计了以二氢噻吩并嘧啶和二氢呋喃并嘧啶为铰链区的先导化合物30和31,与吡咯并嘧啶相比它们具有更高的PKA、一般激酶选择性和更高耐受性。通过对化合物29和30的构效关系分析,得到目标化合物32,其对Akt1和PKA的IC50值分别为1 nM和35 nM,且对PC3-NCI前列腺小鼠肿瘤细胞也具有明显的抑制作用,当以200 mg/kg的每日口服剂量给药时,其显示出肿瘤生长和停滞的剂量依赖性降低 [24]。
GDC0068 (化合物33)是将化合物32与Akt1和蛋白激酶A分子结合后通过X射线晶体衍射分析设计得到的。GDC0068对三种Akt亚型均有明显的抑制效果,其IC50值分别达到5 nM,18 nM,8 nM,可阻断人类癌细胞系中Akt多个下游靶标的磷酸化。GDC0068对Akt的选择性优异,且具有良好的口服暴露可导致对下游生物标记物具有剂量依赖性的药效作用,在异种移植模型中具有强烈的抗肿瘤反应,在该模型中,磷脂酰肌醇3-激酶-Akt-雷帕霉素是哺乳动物的靶标通路被激活 [25]。研究表明,GDC0068是一种新型的,具有高度选择性的ATP竞争性Akt抑制剂,具有令人信服的选择性,功效和口服药代动力学,可单独或与化疗药物一起作为抗癌药支持其临床开发。目前GDC0068正处于临床Ⅲ期的研究试验中,是最有可能上市的Akt抑制剂 [26] (见图9)。

Figure 9. The structure of piperazine compounds (Figure 30~33)
3. 结语与展望
研发以Akt为靶点的小分子抑制剂已经成为抗肿瘤治疗的思路之一,目前已有多个ATP竞争性抑制剂有着良好的生物活性并处于临床研究阶段,如GDC0068、AZD5363、GSK2110183、BAY1125976等。这些抑制剂一般都有着较高的抑制效率,但在选择性和毒副作用等方面仍然有较大的改进空间。相信随着研究地不断开展,会有更多高活性和选择性的Akt抑制剂进入临床研究,在肿瘤的治疗上带来新的突破。
文章引用
王彦凯,黎文海. Akt激酶小分子抑制剂研究进展——ATP竞争性抑制剂研究概述
Research Progress of Small Molecule Inhibitors of Akt Kinase—Overview of ATP Competitive Inhibitor Research[J]. 有机化学研究, 2021, 09(02): 13-21. https://doi.org/10.12677/JOCR.2021.92003
参考文献
- 1. Sale, E.M. and Sale, G.J. (2008) Protein Kinase B: Signalling Roles and Therapeutic Targeting. Cellular and Molecular Life Sciences, 65, Article No. 113. https://doi.org/10.1007/s00018-007-7274-9
- 2. Carnero, A., Blanco-Aparicio, C., Renner, O., Link, W. and Leal, J.F.M. (2008) The PTEN/PI3K/AKT Signalling Pathway in Cancer, Therapeutic Implications. Current Cancer Drug Targets, 8, 187-198. https://doi.org/10.2174/156800908784293659
- 3. Zhou, B.G., Wei, C.S., Zhang, S., Zhang, Z. and Gao, H.M. (2018) Matrine Reversed Multidrug Resistance of Breast Cancer MCF-7/ADR Cells through PI3K/AKT Signaling Pathway. Journal of Cellular Biochemistry, 119, 3885-3891. https://doi.org/10.1002/jcb.26502
- 4. Georgakis, G.V. and Younes, A. (2006) From Rapa Nui to Rapamycin: Targeting PI3K/Akt/mTOR for Cancer Therapy. Expert Review of Anticancer Therapy, 6, 131-140. https://doi.org/10.1586/14737140.6.1.131
- 5. Heerding, D.A., Rhodes, N., Leber, J.D., Clark, T.J., Keenan, R.M., Lafrance, L.V., et al. (2008) Identification of 4-(2-(4-amino-1,2,5-oxadiazol-3-yl)-1-ethyl-7-{[(3S)-3-piperidinylmethyl]oxy}-1H-imidazo[4,5-c]pyridin-4-yl)-2-methyl-3-butyn-2-ol (GSK690693), a Novel Inhibitor of AKT Kinase. Journal of Medicinal Chemistry, 51, 5663-5679. https://doi.org/10.1021/jm8004527
- 6. Rhodes, N., Heerding, D.A., Duckett, D.R., Eberwein, D.J., Knick, V.B., Lansing, T.J., et al. (2008) Characterization of an AKT Kinase Inhibitor with Potent Pharmacodynamic and Antitumor Activity. Cancer Research, 68, 2366-2374. https://doi.org/10.1158/0008-5472.can-07-5783
- 7. Crouthamel, M.-C., Kahana, J.A., Korenchuk, S., Zhang, S.-Y., Sundaresan, G., Eberwein, D.J., et al. (2009) Mechanism and Management of AKT Inhibitor-Induced Hyperglycemia. Clinical Cancer Research, 15, 217-225. https://doi.org/10.1158/1078-0432.CCR-08-1253
- 8. Lippa, B., Pan, G., Corbett, M., Li, C., Kauffman, G.S., Pandit, J., et al. (2008) Synthesis and Structure Based Optimization of Novel Akt Inhibitors. Bioorganic & Medicinal Chemistry Letters, 18, 3359-2263. https://doi.org/10.1016/j.bmcl.2008.04.034
- 9. Freeman-Cook, K.D., Autry, C., Borzillo, G., Gordon, D., Barbacci-Tobin, E., Bernardo, V., et al. (2010) Design of Selective, ATP-Competitive Inhibitors of Akt. Journal of Medicinal Chemistry, 53, 4615-4622. https://doi.org/10.1021/jm1003842
- 10. Caldwell, J.J., Davies, T.G., Donald, A., McHardy, T., Rowlands, M.G., Wynne Aherne, G., et al. (2008) Identification of 4-(4-aminopiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidines as Selective Inhibitors of Protein Kinase B through Fragment Elaboration. Journal of Medicinal Chemistry, 51, 2147-2157. https://doi.org/10.1021/jm701437d
- 11. McHardy, T., Caldwell, J.J., Cheung, K.-M., Hunter, L.J., Taylor, K., Rowlands, M., Ruddle, R., et al. (2010) Discovery of 4-amino-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamides as Selective, Orally Active Inhibitors of Protein Kinase B (Akt). Journal of Medicinal Chemistry, 53, 2239-2249. https://doi.org/10.1021/jm901788j
- 12. Addie, M., Ballard, P., Buttar, D., Crafter, C., Currie, G., Davies, B.R., Debreczeni, J., et al. (2013) Discovery of 4-amino-N-[(1S)-1-(4-chlorophenyl)-3-hydroxypropyl]-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (AZD5363), an Orally Bioavailable, Potent Inhibitor of Akt Kinases. Journal of Medicinal Chemistry, 56, 2059-2073. https://doi.org/10.1021/jm301762v
- 13. Rice, K.D., Kim, M.H., Bussenius, J., Anand, N.K., Blazey, C.M., Bowles, O.J., et al. (2012) Pyrazolopyrimidines as Dual Akt/p70S6K Inhibitors. Bioorganic & Medicinal Chemistry Letters, 22, 2693-2697. https://doi.org/10.1016/j.bmcl.2012.03.011
- 14. Lin, X., Murray, J.M., Rico, A.C., Wang, M.X., Chu, D.T., Zhou, Y., Rosario, M.D., Kaufman, S., Ma, S., Fang, E., Crawford, K. and Jefferson, A.B. (2006) Discovery of 2-pyrimidyl-5-amidothiophenes as Potent Inhibitors for AKT: Synthesis and SAR Studies. Bioorganic & Medicinal Chemistry Letters, 16, 4163-468. https://doi.org/10.1016/j.bmcl.2006.05.092
- 15. Chang, S., Zhang, Z., Zhuang, X., Luo, J., Cao, X., Li, H., Tu, Z., Lu, X., Ren, X. and Ding, K. (2012) New Thiazole Carboxamides as Potent Inhibitors of AKT Kinases. Bioorganic & Medicinal Chemistry Letters, 22, 1208-1212. https://doi.org/10.1016/j.bmcl.2011.11.080
- 16. Zhan, W., Li, D., Che, J., Zhang, L., Yang, B., Hu, Y., Liu, T. and Dong, X. (2014) Integrating Docking Scores, Interaction Profiles and Molecular Descriptors to Improve the Accuracy of Molecular Docking: Toward the Discovery of Novel Akt1 Inhibitors. European Journal of Medicinal Chemistry, 75, 11-20. https://doi.org/10.1016/j.ejmech.2014.01.019
- 17. Saxty, G., Woodhead, S.J., Berdini, V., Davies, T.G., Verdonk, M.L., Wyatt, P.G., et al. (2007) Identification of Inhibitors of Protein Kinase B Using Fragment-Based Lead Discovery. Journal of Medicinal Chemistry, 50, 2293-2296. https://doi.org/10.1021/jm070091b
- 18. Yap, T.A., Walton, M.I., Grimshaw, K.M., te Poele, R.H., Eve, P.D., Valenti, M.R., et al. (2012) AT13148 Is a Novel, Oral Multi-AGC Kinase Inhibitor with Potent Pharmacodynamic and Antitumor Activity. Clinical Cancer Research, 18, 3912-3923. https://doi.org/10.1158/1078-0432.CCR-11-3313
- 19. Dumble, M., Crouthamel, M.-C., Zhang, S.-Y., Schaber, M., Levy, D., Robell, K., et al. (2014) Discovery of Novel AKT Inhibitors with Enhanced Anti-Tumor Effects in Combination with the MEK Inhibitor. PLoS ONE, 9, e100880. https://doi.org/10.1371/journal.pone.0100880
- 20. Tolcher, A.W., Patnaik, A., Papadopoulos, K.P., Rasco, D.W., Becerra, C.R., Allred, A.J., et al. (2015) Phase I study of the MEK Inhibitor Trametinib in Combination with the AKT Inhibitor Afuresertib in Patients with Solid Tumors and Multiple Myeloma. Cancer Chemotherapy and Pharmacology, 75, 183-189. https://doi.org/10.1007/s00280-014-2615-5
- 21. Zhan, W., Xu, L., Dong, X., Dong, J., Yi, X., Ma, X., et al. (2016) Design, Synthesis and Biological Evaluation of Pyrazol-Furan Carboxamide Analogues as Novel Akt Kinase Inhibitors. European Journal of Medicinal Chemistry, 117, 47-58. https://doi.org/10.1016/j.ejmech.2016.03.074
- 22. Dong, X., Zhan, W., Zhao, M., Che, J., Dai, X., Wu, Y., et al. (2019) Discovery of 3,4,6-Trisubstituted Piperidine Derivatives as Orally Active, Low hERG Blocking AKT Inhibitors via Conformational Restriction and Structure-Based Design. Journal of Medicinal Chemistry, 62, 7264-7288. https://doi.org/10.1021/acs.jmedchem.9b00891
- 23. Blake, J.F., Kallan, N.C., Xiao, D., Xu, R., Bencsik, J.R., Skelton, N.J., et al. (2010) Discovery of Pyrrolopyrimidine Inhibitors of AKT. Bioorganic & Medicinal Chemistry Letters, 20, 5607-5612. https://doi.org/10.1016/j.bmcl.2010.08.053
- 24. Bencsik, J.R., Xiao, D., Blake, J.F., Kallan, N.C., Mitchell, I.S., Spencer, K.L., et al. (2010) Discovery of Dihydrothieno- and Dihydrofuropyrimidines as Potent Pan AKT Inhibitors. Bioorganic & Medicinal Chemistry Letters, 20, 7037-7041. https://doi.org/10.1016/j.bmcl.2010.09.112
- 25. Blake, J.F., Xu, R., Bencsik, J.R., Xiao, D., Kallan, N.C., Schlachter, S., et al. (2012) Discovery and Preclinical Pharmacology of a Selective ATP-Competitive AKT Inhibitor (GDC-0068) for the Treatment of Human Tumors. Journal of Medicinal Chemistry, 55, 8110-8127. https://doi.org/10.1021/jm301024w
- 26. Lin, J., Sampath, D., Nannini, M.A., Lee, B.B., Degtyarev, M., Oeh, J., et al. (2013) Targeting Activated AKT with GDC-0068, a Novel Selective AKT Inhibitor That Is Efficacious in Multiple Tumor Models. Clinical Cancer Research, 19, 1760-1772. https://doi.org/10.1158/1078-0432.CCR-12-3072