Applied Physics
Vol.08 No.02(2018), Article ID:23796,10
pages
10.12677/APP.2018.82012
First-Principles Study on Properties of Ni-SiCNP
Yan Xu1, Zhenbo Zhu1,2, Rengang Song1, Xiaolu Zhu3, Canglong Wang3
1College of Electronic, Communication and Physics, Shandong University of Science & Technology, Qingdao Shandong
2Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai
3Institute of Modern Physics, Chinese Academy of Sciences, Lanzhou Gansu

Received: Feb. 3rd, 2018; accepted: Feb. 19th, 2018; published: Feb. 26th, 2018

ABSTRACT
For Ni-SiCNP, the experimental results show that nano-silicon carbide uniformly dispersed manner can be employed to enhance the mechanical properties and anti-irradiation properties. In this paper, the mechanical properties and the formation energy of helium defects under irradiation conditions are calculated by the density functional theory of first-principles, and obtain the mechanical properties of the composites with the change of the mass fraction of silicon carbide. Further analysis shows that the interface is easier to capture helium Atomic and silicon carbide particles can enhance the mechanical properties of pure nickel. The results provide a theoretical basis for the development of nickel-based silicon carbide composites.
Keywords:Ni-SiCNP, First Principle, Helium Defect Formation Energy, Mechanical Properties
镍基碳化硅复合材料结构性能 第一性原理研究
徐岩1,朱振博1,2,宋仁刚1,朱晓禄3,王苍龙3
1山东科技大学电子通信与物理学院,山东 青岛
2中国科学院上海应用物理研究所,上海
3中国科学院近代物理研究所散裂靶研究室,甘肃 兰州
收稿日期:2018年2月3日;录用日期:2018年2月19日;发布日期:2018年2月26日

摘 要
实验结果表明:对于镍基碳化硅复合材料,可以采用纳米碳化硅均匀弥散的方式来增强其力学性能和抗辐照性能。本文通过密度泛函理论对该复合材料辐照条件下的氦缺陷形成能和力学性能进行了第一性原理计算,结果表明镍基和碳化硅颗粒界面处更易捕获氦原子,且碳化硅颗粒会改善纯镍的力学性质,进一步分析得到该复合材料力学性质随碳化硅质量分数变化的曲线,研究结果为促进镍基碳化硅复合材料的发展提供理论基础。
关键词 :镍基碳化硅复合材料,第一性原理,氦缺陷形成能,力学性质

Copyright © 2018 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 引言
先进钍基熔盐 [1] 堆作为唯一的液态燃料堆展示出独特的优点,但其对堆材料的选择也提出了更高的要求。最优备选材料哈氏合金(Hastelloy N) [2] 具有优异的耐氟化物熔盐腐蚀性能,但存在一定的局限性:1) Hastelloy N合金的最高许用温度为704℃;2) 镍的中子吸收截面大,易与快中子发生
为了满足抗辐照结构材料的需求,上海应用物理研究所借鉴氧化物弥散强化(Oxide Dispersion Strengthened, ODS)铁素体钢中氧化物弥散颗粒加强的方法,制备出纳米碳化硅均匀弥散的镍基碳化硅复合材料。新材料采用结构单一成分的镍为基体,具有优异高温力学性能、耐氟化物熔盐腐蚀性及抗辐照性能的碳化硅为弥散相,并通过实验证明了纯镍中弥散的碳化硅颗粒能有效的抑制辐照过程中氦泡的生长,从而改进了材料的抗氦脆及肿胀性能 [4] 。为了得到更好的材料性能,杨超等人 [5] 对其不同碳化硅质量下的力学性能进行了研究,得到了屈服强度、抗张强度和维氏硬度随碳化硅质量分数变化曲线。
实验已经证明纳米颗粒对金属基材料多方面性质起到加强作用 [6] [7] ,但对于颗粒增强材料的第一性原理计算较少。本文运用第一性原理方法开展了对镍基碳化硅复合材料的研究,对四个位置掺杂氦原子后氦缺陷形成能计算,得到氦原子分布概率最大的位置;对掺杂氦后的界面键合状况分析,初步解释了纳米碳化硅颗粒在镍基体中的抗氦脆机理。同时,也对材料的力学性质进行了研究,得到了杨氏模量、体积模量、剪切模量对碳化硅质量分数的变化曲线,为寻找镍基碳化硅复合材料的最佳纳米碳化硅颗粒弥散比例提供了线索。另外,本文模型的建立对研究颗粒增强复合材料提供了新方法。
2. 计算方法和模型
本文所有的计算均采用Materials Studio软件中的CASTEP模块完成。鉴于过渡金属Ni的电子组态为[Ar]4s23d8,C的电子组态为[He]2s22p2,Si的电子组态为[Ne]3s23p1,交换关联能泛函选用局域密度近似(local-density approximation, LDA) [8] ,赝势场选用超软赝势(ultra-soft pseudopotential, USPP) [9] 。平面波的截断能为340 eV,布里渊区K点取值为8 × 8 × 1,体系每个原子的总能量收敛值取5.0 × 10−6 eV,每个原子上的力低于0.01 eV/Å,公差偏移小于5.0 × 10−4 Å,应力偏差小于0.02 Gpa。
理想镍晶体和理想碳化硅晶体的结构均为面心立方,取两晶体(100面)匹配测试(晶格错配率小于5%)后,建立所需模型。该模型选用2 × 2 × 3碳化硅超胞和2 × 2 × 2镍超胞构建;采用夹层结构表示周期结构;采用固定碳化硅中间层方式表示碳化硅颗粒。经过充分驰豫后的晶格如图1。晶格常数为a = 2.99Å,b = 2.99Å,c = 22.71Å,属于PMM2晶系。周期结构产生两个界面:C-Si-Ni界面为Si原子与Ni原子相邻,如图1下界面;Si-C-Ni界面为C原子与Ni原子相邻,如图1上界面。C-Si-Ni界面间距为1.14 Å,Si-C-Ni界面间距为1.10 Å,界面处存在C-Si-Ni和Si-C-Ni原子链 [10] ,维系着界面的稳定。
3. 计算结果与讨论
3.1. 氦缺陷形成能
晶体中氦的行为决定了氦原子在不同位置的能量状态,根据玻尔兹曼分布,能量最低的位置其占据概率越大 [11] 。为了更好的得到材料捕获氦原子概率最大的位置,我们取四个间隙位置,通过计算氦缺陷形成能来验证碳化硅和镍界面处有较好捕获氦的能力。如图2所示,A点位于C-Si-Ni界面间隙内,B点位于Si-C-Ni界面间隙内,C点、D点分别位于C-Si-Ni界面和Si-C-Ni界面相邻界面的镍间隙内。经过充分的驰豫优化,得到系统的最优结构和总能。
氦缺陷形成能计算公式如下 [12] :
(1)
其中, 、 和 分别为含氦系统总能、完美Ni-SiCNP的能量和氦原子的化学势,计算结果如下表:
由表1可知,B位置的氦缺陷形成能最低,其次为A位置。当氦原子掺入C位置时,会使得镍原子金属键破坏,不能稳定存在。其余三个位置体积变化较小,且氦缺陷形成能差距较小,都可能在材料中稳定存在,且在B位置存在的概率更大。一般情况下,晶格体积变化越小,则系统越稳定,但对于氦原子掺入系统有些不同 [8] 。由体积变化可知,B位置处镍晶格显示出轻微的变形,这个变形将留下氦间隙原子的空间,从而导致总能量的大量下降,使得氦缺陷形成能最低 [9] 。

Figure 1. Optimized cell structure
图1. 优化后晶胞图

Figure 2. Helium atom position distribution
图2. 氦原子位置分布图

Table 1. Comparison of defect formation helium and unit cell volume change
表1. 氦缺陷形成能比较和晶胞体积变化
3.2. 界面成键分析
为了更加清晰从键合能力来观察氦的存在对界面的影响,我们对界面处做态密度图和布局分析,见图3。态密度表示单位能量范围内所允许的电子数,也就是说电子在某一能量范围的分布情况,能反应出电子在各个轨道的分布情况,反映出原子与原子之间的相互作用情况,并且还可以揭示化学键的信息。P值代表布局数。布局数是指电子在原子轨道上的分布,分析这种布局,有助于了解分子中原子的成键情况。Ni与Si、C原子的杂化主要体现在Ni的d电子与Si、C的s、p电子轨道杂化上,因此只将Ni的d电子态密度分析图画出。键的布局数表示两个成键原子之间电子云的重叠程度,通常用来判断化学成键的共价性强弱:正值越大,则共价键越强 [13] [14] [15] 。
对比Si-C-Ni界面分波态密度分析图:He掺入之前,Ni的d态与C的p态在−4 eV、0 eV出现两个峰值;He掺入之后,Ni的d态更加局域化,在0 eV处态密度值更高,且C的p态在此处的密度值也升高,即Ni的d态与C的p态杂化更强。反映在布局分析,表现在C-Ni的P值的增加,共价键更强,在一定程度上弥补了He的掺入使得界面结合力降低。
对比C-Si-Ni界面分波态密度分析图:He掺入之前,Ni(1)和Ni(2)的d态与Si的p态在−5 eV附近出现重叠峰。Ni(1)的d态与Si的p态和C的p态在−1 eV附近出现重叠峰;He掺入之后,Ni(1)的d态峰值降低,且在−1 eV不在与C的p态存在重叠峰。Ni(2)的d态峰值升高。结合布局分析,Si--Ni(1)的P值减小,则Si-Ni键的结合力减弱。C与Ni(1)之间不再形成共价键,这是由于He的掺入使得C-Ni(1)键变长,不再成键。Si--Ni(2)的P值增大,即Si与Ni(2)的结合力增强,在一定程度上弥补了He的掺入使得界面结合力降低 [11] [14] 。
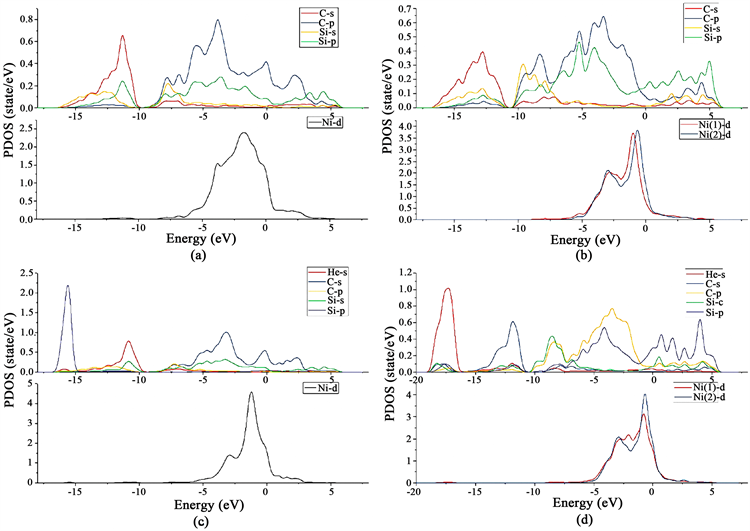
Figure 3. (a) Interface state density of pure Si-C-Ni; (b) Interface state density of pure C-Si-Ni; (c) Interface state density of He in Si-C-Ni; (d) Interface state density of He in C-Si-Ni
图3. (a) 纯Si-C-Ni界面分波态密度图;(b) 纯C-Si-Ni界面分波态密度图;(c) He in Si-C-Ni界面分波态密度图;(d) He in C-Si-Ni界面分波态密度图
由表2可知,在Si-C-Ni界面,He--Ni的P值为1.07;在C-Si-Ni界面,He--Ni(1)的P值为1.43。由布局分析的性质可知,在C-Si-Ni界面He与Ni的成键更强。若He原子与金属原子的相互作用力较强,则He对金属原子晶界的影响越大,即He与Ni所形成的共价键更强,则Ni与界面处的C和Si形成的共价键变弱,则界面更不稳定。He的掺入使得C-Si-Ni界面比Si-C-Ni界面更不稳定,其氦缺陷形成能更大,符合缺陷形成能模拟所得。
3.3. 单个氦原子迁移
氦原子作为嬗变产物,在镍基中不易被溶解,具有较高的迁移能力,会自捕获或者在晶界、相界、空位等处被捕获成团。氦原子从一个位置迁移到另一个位置,需要跨过一个因基体原子周期性势场存在而引起的势垒高度,此势垒表示迁移激活能的大小 [11] [12] 。为了研究氦原子在镍基碳化硅复合材料中的迁移扩散行为,我们计算了单个在镍基碳化硅复合材料不同占据位置氦缺陷形成能的结果,且孤立氦原子主要占据能量最低的位置,也就是Si-C-Ni界面处,即B位置。考虑到氦原子会在最优先迁移到最稳定位置,因此我们主要计算了氦原子在两个稳定位置之间的迁移行为 [11] 。我们主要计算了两条路径,如图4所示:路径(1)氦原子从B位置经过镍间隙,迁移到B位置;路径(2)氦原子从B位置先迁移到Ni-C-Si间隙处,然后迁移到B位置。
计算结果见图5,经过对比可知,氦原子在Ni-SiCNP材料中优先选择路径(1)进行迁移,即先迁移到
Table 2. The layout of the keys interface analysis
表2. 界面处各键单位键长的重叠布局

Figure 4. (a) Path 1; (b) Path 2
图4. (a) 路径(1);(b) 路径(2)

Figure 5. Barrier of path 1; Barrier of path 2
图5. 路径(1)势垒;路径(2)势垒
镍间隙,后迁移到B位置,相应的迁移势能为0.6 eV。路径(2)的迁移势能为0.9 eV左右,与路径(1)迁移势垒相差较小,也有从此路径迁移的可能性。
3.4. 力学性质
对于第二相加强材料而言,其第二相颗粒的质量分数对于材料的力学性质有着决定作用 [12] 。因此,找到最恰当的第二相颗粒的质量分数对于材料更广泛应用具有重大意义。
若忽略温度对体系总能的影响,体系总能 是体积 和应变 的函数,在应变比较小的情况下,弹性常数可以通过应变后体系总能函数的泰勒级数展开得到(忽略的 更高次相) [16] :
(2)
式中 是晶格形变前时的体积, 是形变前体系总能。对于正交晶系来说,具有10个独立弹性系数,分别为:C11,C22,C33,C44,C55,C66,C12,C13,C23。
对于正交晶系,体积模量B和剪切模量G的计算方法为 [17] :
(3)
其中,BV和GV为采用Voigt模型所得,BR和GR为采用Reuss模型所得,GH和BH为Hill模型所得。Hill模型为Voigt模型和Reuss模型所求的算数平均值,大量实验证明,Hill模型与实际测量更为接近 [18] 。(Sij为Cij的逆矩阵)
根据弹性力学的性质可知,弹性模量(E)和泊松系数(μ)的计算方法为:
(4)
对于正交晶系而言,固体的机械稳定性可以通过下面的公式来判断 [19] :
(5)
本文通过固定碳化硅中间层的方法表示碳化硅为纳米颗粒存在,能够更好的模拟出纳米颗粒均匀分散在镍基中的材料性能:在真实复合材料中,碳化硅颗粒内部结构不会发生形变。表3列出了1到6层镍基复合材料和纯镍(表中用∞表示)的体积模量(B)、剪切模量(G)、B/G、杨氏模量(E)、泊松系数(μ)。碳化硅颗粒为高硬度材料,作为第二相对材料有明显的强化作用,复合材料最大体积模量、剪切模量和弹性模量分别为纯镍的1.32、1.64和1.59倍。
观察表3,镍层数由1到4层时,其体积模量(G)是随之变大,到达极大值为233;在镍层数为5、6层时,有所下降,并且都大于纯镍的体积模量,且极大值为纯镍的1.3倍。体积模量与晶体的结合能或键能紧密相关,通常为评判平均键能强度的标准,体积模量越大则说明相应的平均价键强度越高。根据Pugh经验判断 [20] [21] ,B/G一般用来分析材料的脆性和延展性。对于六方和立方对称性的金属化合物,通常采用1.75作为区分延展性材料和脆性材料的标准。B/G低于1.75的材料为脆性材料,反之为延展性材料。超过两层镍时,其B/G基本不发生变化。泊松系数与B/G有相同的变化规律。泊松系数与单向变形所引起垂直于该方向的另外两个方向变化量有关,较高的泊松系数比说明较小的体积变。则镍层数的增加对其延展性和单向变形所引起的体积变化基本没有影响 [21] 。
为了更加清晰的观察弹性模量(B)、剪切模量(G)和杨氏模量(E)的变化趋势,作图6,可以观察到B、G、E都存在随着碳化硅质量分数的增多先增多后变小的趋势。图7为实验得到的维氏硬度、屈服度、抗张强度曲线,都存在随着碳化硅质量分数的增多先增多后减少的趋势,因此认为模型建立与实验所得较为符合。



Figure 6. Bulk modulus, shear modulus, Young’s modulus change with the number of nickel-based layers
图6. 体积模量(B)、剪切模量(G)、杨氏模量(E)随镍基层数变化



Figure 7. Tensile strength, Yield strength, Vickers hardness with the silicon carbide mass fraction changes [5]
图7. 抗张强度,屈服度,维氏硬度随碳化硅质量分数变化 [5]
Table 3. Bulk modulus, the shear modulus, B/G, Young’s modulus, Poisson’s ratio at different numbers nickel-based layer
表3. 不同镍基层数下的体积模量、剪切模量、B/G、杨氏模量、泊松比
4. 结论
本文利用密度泛函理论,对镍基碳化硅复合材料的模型构造、氦缺陷形成能、力学性质进行相关求解和计算,并结合现有理论和实验数据对模型现象进行了理论分析。
1) 对材料特定位置缺陷形成能的计算可知,Si-C-Ni界面处氦缺陷形成能最低,能较好的捕获氦原子。
2) 通过对界面处态密度分析和布局分析可知,氦原子会优先与镍原子成键,从而对界面的结合进行破坏。相比Si-C-Ni界面,氦原子对C-Si-Ni界面的键合破坏更严重。
3) 对不同碳化硅质量分数复合材料的力学性质的计算,随着碳化硅质量分数的增加,其体积模量、剪切模量、弹性模量都有先增加后减少的趋势,且其变化趋势与实验所得相关力学性质基本相同,在一定程度上证明了模型的正确性。
基金项目
国家自然科学基金(项目编号:11547037,11304324和11604181)和中国科学院战略性先导科技专项(项目编号:XDA03030100)资助课题。
文章引用
徐 岩,朱振博,宋仁刚,朱晓禄,王苍龙. 镍基碳化硅复合材料结构性能第一性原理研究
First-Principles Study on Properties of Ni-SiCNP[J]. 应用物理, 2018, 08(02): 100-109. http://dx.doi.org/10.12677/APP.2018.82012
参考文献 (References)
- 1. 江绵恒, 徐洪杰, 戴志敏. 未来先进核裂变能——TMSR核能系统[J]. 中国科学院院刊, 2012, 27(3): 366-374.
- 2. 林建波. Hastelloy N合金的离子辐照损伤及辐照后熔盐腐蚀机理研究[D]: [博士学位论文]. 北京: 中国科学院大学, 2014.
- 3. Angeliu, T.M/, Ward, J.T. and Witter, J.K. (2007) Assessing the Effects of Radiation Damage on Ni-Base Alloys for the Prometheus Space Reactor System. Journal of Nuclear Materials, 366, 223-237. https://doi.org/10.1016/j.jnucmat.2007.01.217
- 4. 周晓苓. 镍基碳化硅复合材料的辐照性能研究[D]: [硕士学位论文]. 上海: 中国科学院研究生院(上海应用物理研究所), 2016.
- 5. Yang, C., Huang, H., Thorogood, G.J., et al. (2016) The Effect of Grain Size and Dislocation Density on the Tensile Properties of Ni-SiCNP, Composites during Annealing. Journal of Materials Engineering & Performance, 25, 726-733. https://doi.org/10.1007/s11665-016-1938-2
- 6. 樊建中, 姚忠凯, 李义春, 等. 颗粒增强铝基复合材料研究进展[J]. 材料导报, 1997(3): 48-51.
- 7. 贺毅强. 颗粒增强金属基复合材料的研究进展[J]. 热加工工艺, 2012, 41(2): 133-136.
- 8. Probert, D.M. (2006) Electronic Structure—Basic Theory and Practical Methods [Book Review]. IEEE Circuits & Devices Magazine, 22, 34-34. https://doi.org/10.1109/MCD.2006.1708382
- 9. Huang, H.F., Zhang, W., Reyes, M.D.L., et al. (2016) Mitigation of He Embrittlement and Swelling in Nickel by Dispersed SiC Nanoparticles. Materials & Design, 90, 359-363.
- 10. Profeta, G., Continenza, A. and Freeman, A.J. (2001) Energetics and Bonding Properties of the Ni/β-SiC (001) Interface: An ab-initio Study. Physical Review B: Condensed Matter, 64, 045303. https://doi.org/10.1103/PhysRevB.64.045303
- 11. 贾丽霞. 核结构材料中氢氦行为的第一性原理研究[D]: [硕士学位论文]. 上海: 复旦大学, 2013.
- 12. 姚宝殿, 胡桂青, 于治水, 等. H, He对TiSiC材料力学性能影响的第一性原理研究[J]. 物理学报, 2016, 65(2): 026202.
- 13. 唐杰. Fe-Cr-Al合金界面强化机理的第一性原理研究[D]: [硕士学位论文]. 沈阳: 沈阳师范大学, 2015.
- 14. 许红斌, 王渊旭. 过渡金属Tc及其氮化物TcN, TcN2, TcN3与TcN4低压缩性的第一性原理计算研究[J]. 物理学报, 2009, 58(8): 5645-5652.
- 15. 潘勇, 管伟明, 陈松, 等. Pt(100)/ZrO2(100)界面性质的第一性原理[J]. 稀有金属材料与工程, 2011(1): 80-84.
- 16. 唐杰, 张国英, 鲍君善, 等. 杂质S对Fe/Al2O3界面结合影响的第一性原理研究[J]. 物理学报, 2014, 63(18): 401-406.
- 17. 汝强, 胡社军, 赵灵智. LiFePO(=0.0, 0.75, 1.0)电子结构与弹性性质的第一性原理研究[J]. 物理学报, 2011, 60(3): 448-457.
- 18. Wu, Z.J., Zhao, E.J., Xiang, H.P., et al. (2007) Publisher’s Note: Crystal Structures and Elastic Properties of Superhard IrN[sub 2] and IrN[sub 3] from First Principles. Physical Review B, 76, ArticleID: 059904.
- 19. 王斌, 刘颖, 叶金文. 高压下TiC的弹性、电子结构及热力学性质的第一性原理计算[J]. 物理学报, 2012, 61(18): 186501.
- 20. Hayatullah, Murtaza, G., Khenata, R., et al. (2013) First Principle Calculations of the Ground and Excited State Properties of RbPbF3. Chinese Physics Letters, 30, 116-121. https://doi.org/10.1088/0256-307X/30/9/097101
- 21. 王云江. 镍基合金力学性能与元素分配行为的第一原理研究[D]: [博士学位论文]. 北京: 清华大学, 2010.