Material Sciences
Vol.
13
No.
08
(
2023
), Article ID:
70136
,
9
pages
10.12677/MS.2023.138079
MoS2/WS2超晶格电子性质的研究
王金华*,聂亚婷
天津职业技术师范大学理学院,天津
收稿日期:2023年7月4日;录用日期:2023年7月28日;发布日期:2023年8月7日

摘要
通过基于密度泛函理论的第一性原理方法,研究了MoS2/WS2超晶格纳米带电子结构及双轴拉伸应力、压缩应力对其电子性质的影响。研究结果显示:MoS2/WS2是直接带隙半导体,扶手型MoS2/WS2纳米带也为直接带隙半导体。对MoS2/WS2超晶格纳米带施加双轴拉伸应力后其带隙随着应力的增加而增加,施加压缩应力后带隙宽度随应力的增加而减小。通过对能带结构和分波态密度分析得到,费米能级附近的能态主要由Mo、W原子的d轨道和S原子的p轨道贡献。
关键词
超晶格,应力,第一性原理,能带结构

Study on Electronic Properties of MoS2/WS2 Superlattice
Jinhua Wang*, Yating Nie
School of Science, Tianjin University of Technology and Education, Tianjin
Received: Jul. 4th, 2023; accepted: Jul. 28th, 2023; published: Aug. 7th, 2023

ABSTRACT
The electronic structure of MoS2/WS2 Superlattice nanoribbons and the effects of biaxial tensile stress and compressive stress on their electronic properties were studied by the first principle method based on Density functional theory. The calculated results indicate that the MoS2/WS2 superlattice is a direct bandgap semiconductor material, and armchair MoS2/WS2 nanoribbon is also direct band gap semiconductor material. The band gap of MoS2/WS2 superlattice nanoribbons increases with the increase of biaxial tensile stress, while the band gap width decreases with the increase of compressive stress. Through the analysis of band structure and partial density of states, it is found that the energy states near Fermi level are mainly contributed by the d orbitals Mo and W atoms and the p orbitals of S atoms.
Keywords:Superlattice, Strain, First-Principles, Band Structure

Copyright © 2023 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/


1. 引言
随着半导体工艺的发展以及硅工艺的发展到达极限,二维材料逐渐进入人们的视野。Novoselov和Geim [1] 利用机械剥离法从石墨烯中剥离出二维石墨烯后,二维材料的研究成为科学家们的研究热点。最早发现的石墨烯在光、电、热、力学等方面具备优异的性质,如良好的韧性,载流子迁移率等。但是,石墨烯的零带隙使得其很难具有器件的开关状态,故石墨烯在光电子应用中受到限制。因此,类石墨烯的二维半导体材料成为研究热点。过渡金属硫化物(TMDCs)是典型的半导体材料,具有类似石墨烯的结构,由六方晶系的原子单层或少层组成的层状结构,层内原子由强共价键相互连接,层间由弱范德华力连接,且具有近乎完美的带隙宽度,它们的带隙约为1~2 eV [2] - [7] 。二维过渡金属硫化物(TMDCs)有许多适用于热电光电材料的特殊性质。MoS2因其化学稳定性好以及比表面积和表面活性大,被广泛应用于电化学析氢以及固体润滑剂等领域。
通过施加外电场、应力等可以对材料的性质进行调控,进而扩大其应用领域 [8] - [15] 。外加电场可有效的调节能带结构,Ramasubramaniam等人 [11] 研究发现随着外加电场的增加,双层过渡金属硫化物材料的带隙单调减少,发生半导体到金属的转变。通过化学方法调节能带结构,往往会使原子膜被破坏,进而导致无序性。机械应力可以有效地调节二维过渡金属硫化物TMDCs材料的能带结构,许多实验和理论研究了应力对能带结构的调控。比如,Yun [12] 等人应用第一性原理方法研究应力对单层MX2电子结构的影响,拉伸应力使能隙减小,压缩应力使带隙增大。随着拉伸应力的增加单层MX2会发生半导体到金属的转变。Johari和Shenoy [13] 等研究发现应力能改变半导体过渡金属硫化物的带隙,发生半导体到金属的转变,这些变化很大程度上取决于施加应变的类型和硫族化物原子的类型。Scalise [14] 等人研究了对单层和双层MoS2施加双轴应力时其电子结构的变化,张应力和压应力对能带结构有不同的影响,随着张应力增大带隙减小,随着压应力增大,带隙先增加然后减小,单层和双层MoS2最终由半导体变为金属。组建异质结也是能带调控的方法之一。Kośmider和Fernández-Rossier [15] 等人研究了MoS2-WS2双层异质结材料,研究发现双层异质结具有带隙,且带隙小于单层MoS2、WS2材料的带隙。
本文将通过第一性原理方法研究MoS2/WS2超晶格的能带结构及应力对MoS2/WS2超晶格结构的调控,进一步分析施加不同应力时电子结构变化的根本原因。
2. 计算方法和模型
采用基于密度泛函理论的Quantum Espresso (QE)软件包进行计算,运用广义梯度近似(GGA)的Perdew Burke Ernzerhof (PBE)方法计算电子间的交换关联能。结构优化前进行收敛性测试,其中能量收敛标准为1.0 × e−10 Ry,平面波截断能为60 Ry,原子间相互作用力收敛标准为1 × 10−3 Ry/au。MoS2/WS2超晶格布里渊区的积分计算采用k点网格为8 × 4 × 1。为防止因周期性计算方法而引入相互作用,故在其表面加超过12 Å的真空层,计算了体系的几何结构,分析了其电子性质。通过改变晶格常数实现对MoS2/WS2超晶格施加应力。施加应力时产生的应变为
上式中,a代表未施加压力时的晶格常数, 代表施加应力与未施加应力时的晶格常数之差。施加拉伸应力时, ;施加压缩应力时, 。
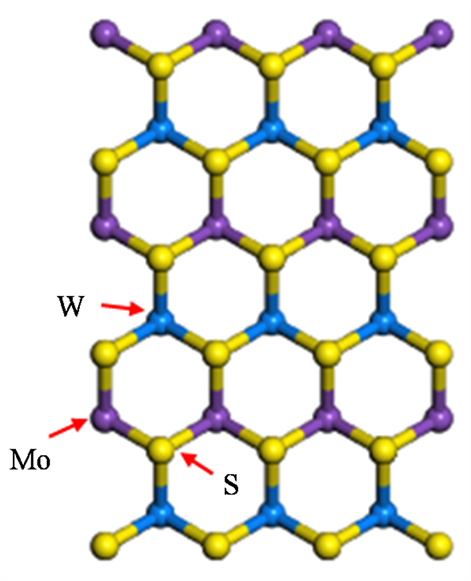
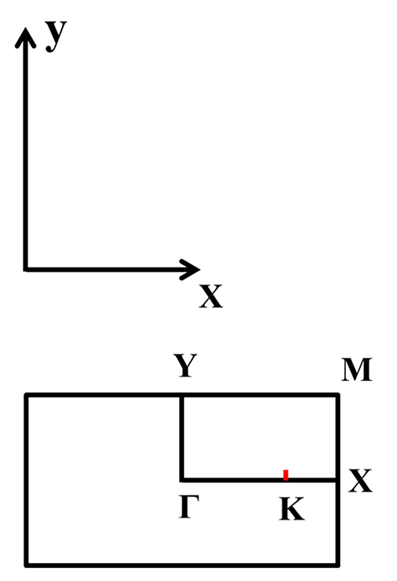
根据不同材料导电性的不同,异质结可以分为同型异质结和异型异质结。原子间距以及晶体结构相似是组成异质结的重要条件。MoS2和WS2的晶体结构相同,带隙不同,晶格失配较小,可以构成异质超晶格。首先通过计算得到晶格常数为3.18 Å时,MoS2、WS2体系能量最低。因MoS2和WS2具有相同的结构,晶格常数又相同,所以选取晶格常数为3.18 Å的MoS2单层与WS2单层构建超晶格结构如图1所示,其中紫色为钼原子,蓝色为钨原子,黄色为硫原子。布里渊区中高对称点Γ、K、X、Y、M已被标出。
 (a)
(a)
 (b)
(b)
 (c)
(c)
Figure 1. The structure of MoS2/WS2 superlattice. (a) Front view; (b) Side view; (c) High symmetry point of the first Brillouin zone
图1. MoS2/WS2超晶格结构。(a) 正视图;(b) 侧视图;(c) 第一布里渊区高对称点
3. 结果和讨论
3.1. MoS2/WS2超晶格能带结构
施加应力是调节材料能带结构的最好的方法之一。与化学方法相比,应力不会破坏材料的结构,对其性质影响较小。相比于施加外电场,应力对材料能带结构的影响较大。本文将研究应力对单层MoS2/WS2超晶格电子结构的影响。
通过能带结构的计算,得到MoS2/WS2超晶格的能带图,如下图2所示。从图2可发现MoS2/WS2超晶格的带隙为直接带隙,带隙宽度为1.724 eV。其价带顶(VBM)和导带底(CBM)在同一点,且位于高对称点K点位置。相较于Γ-X方向,Γ-Y方向能带曲线较为平缓,成键强度小。沿Γ-X方向,能带曲线较为发散,K点带隙宽度最小,那么成键强度也最强。
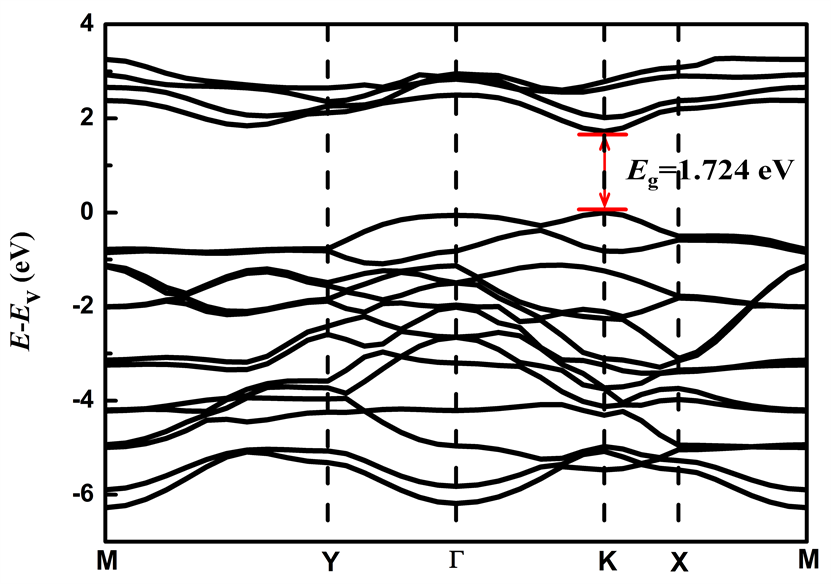
Figure 2. The energy band structure of MoS2/WS2 superlattice
图2. MoS2/WS2超晶格的能带结构
3.2. 扶手型MoS2/WS2纳米带模型建立与参数选取
通过切割横向MoS2/WS2单层超晶格得到纳米带,存在具有不同手性和边缘结构的锯齿型(zz-NR)和扶手椅型(ac-NR)纳米带。本文中研究扶手椅型的MoS2/WS2纳米带,沿宽度方向对其两端的原子固定。其次对构建的纳米带结构进行优化,布里渊区的积分计算采用k点网格为 8 × 2 × 1,体系的截断能选为60 Ry,选取的真空层大于12 Å。
MoS2/WS2纳米带的结构图模型如下图3所示,其中紫色为钼原子,蓝色为钨原子,黄色为硫原子。
 (a)
(a)
 (b)
(b)
Figure 3. The structure of MoS2/WS2 nanoribbon. (a) Front view; (b) High symmetry point of the first Brillouin zone
图3. MoS2/WS2纳米带结构。(a) 正视图;(b) 第一布里渊区高对称点
3.2.1. 本征MoS2/WS2纳米带电子结构分析
图4为未施加应力时MoS2/WS2纳米带的能带结构和态密度图。从图4可知未施加应力时,本征MoS2/WS2纳米带价带顶(VBM)与导带底(CBM)位于Γ点,其带隙宽度为0.537 eV,为直接带隙半导体材料。
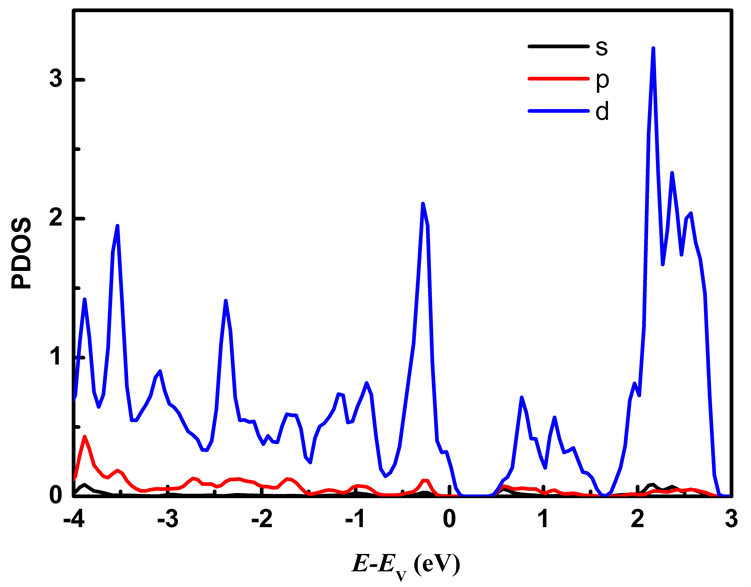
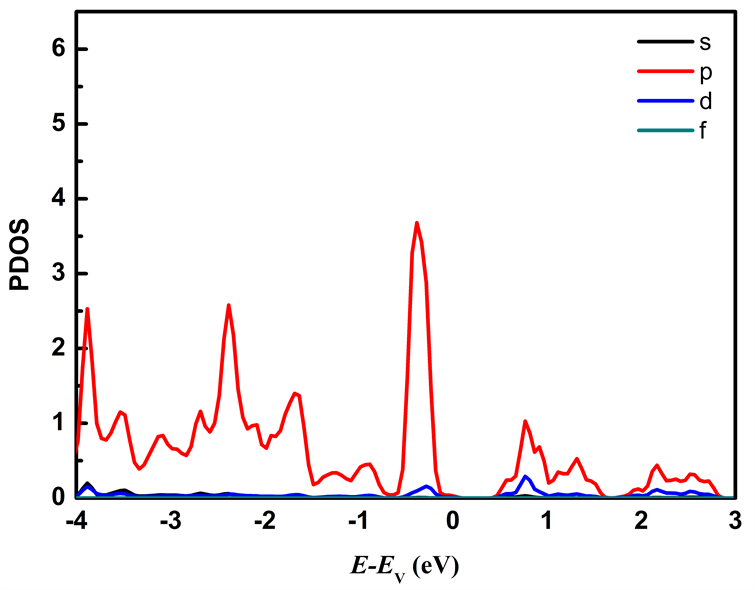
图5为本征MoS2/WS2纳米带的分波态密度图,本征MoS2/WS2纳米带费米能级附近的电子态主要有Mo原子的d轨道、W原子d轨道以及S原子的p轨道。从图中可得出价带顶主要由Mo原子的d轨道、W原子的d轨道和S原子的p轨道构成,导带底主要由Mo原子的d轨道贡献。态密度的结果与能带结构的结果吻合,与单层MoS2/WS2超晶格相比,MoS2/WS2纳米带的带隙明显减小,电子更容易发生跃迁,导电性增强,同时也能提高吸光性能。
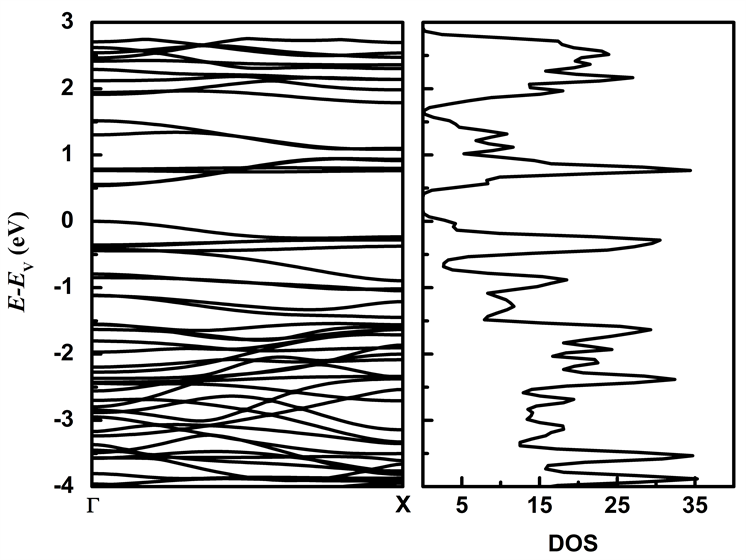
Figure 4. The energy band structure and density of state of MoS2/WS2 nanoribbion
图4. MoS2/WS2纳米带能带结构和态密度
 (a)
(a)
 (b)
(b)
 (c)
(c)
Figure 5. The partial density of states of MoS2/WS2 nanoribbion. (a) Partial density of states of Mo; (b) Partial density of states of W; (c) Partial density of states of S
图5. MoS2/WS2纳米带的分波态密度。(a) Mo的分波态密度;(b) W的分波态密度;(c) S的分波态密度
3.2.2. 应力对MoS2/WS2纳米带电子结构的影响
对二维材料施加应力是能带调控最有效的调控方式。文中研究了对MoS2/WS2纳米带施加不同拉伸和压缩应力后能带结构以及态密度的变化情况。本文对纳米带施加双轴压缩应力,相应的应变为ε = −1%和ε = −2%,同样对纳米带施加双轴拉伸应力,相应的应变为ε = 1%、ε = 2%、ε = 3%。图6所示为不同应力下MoS2/WS2纳米带带隙宽度的变化曲线。MoS2/WS2纳米带在未施加应力时带隙宽度为0.537 eV。从图6中可以得出,在研究范围内,对MoS2/WS2纳米带施加压缩应力时,随着应力的增加,带隙宽度逐渐减小;施加拉伸应力时,随着应力的增加,带隙宽度增大。当系统受到压缩或拉伸应力时,原子的相对位置发生改变,从而影响成键性能以及不同轨道之间的耦合,使导带底和价带顶附近的电子杂化程度发生改变,进而引起能带结构的变化。
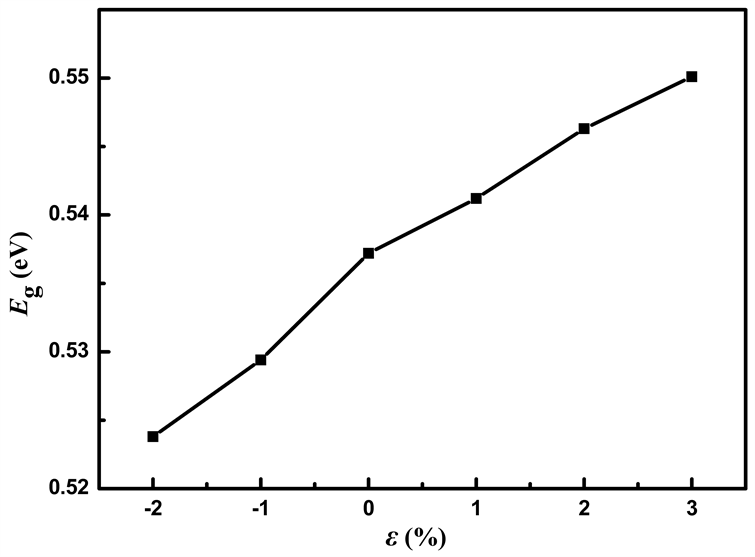
Figure 6. The variation of bandgap width with stress of MoS2/WS2 nanoribbion
图6. MoS2/WS2纳米带的带隙宽度随应力的变化
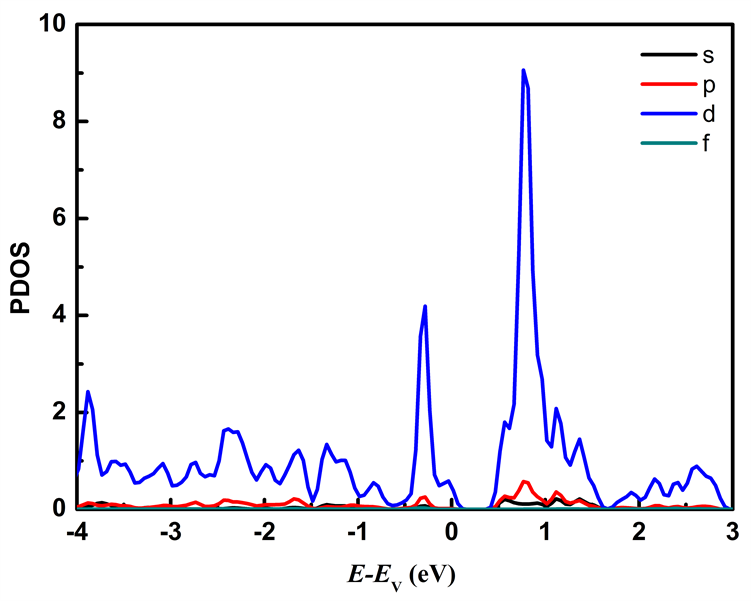
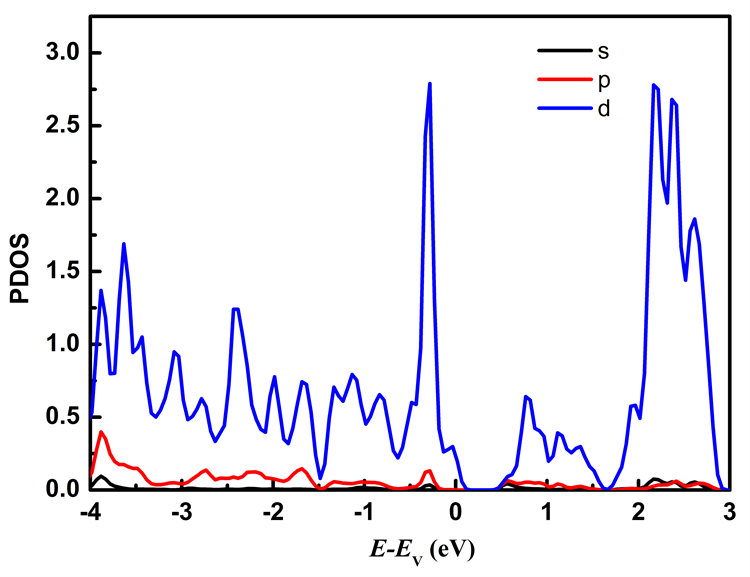
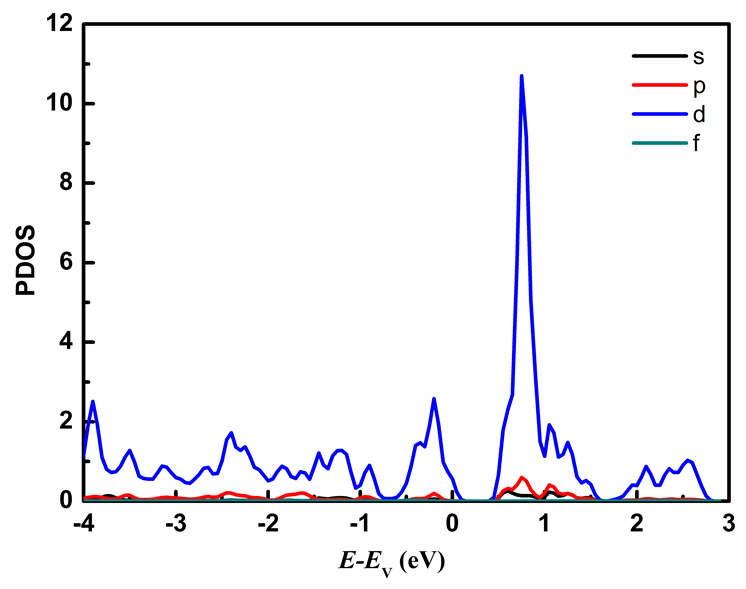
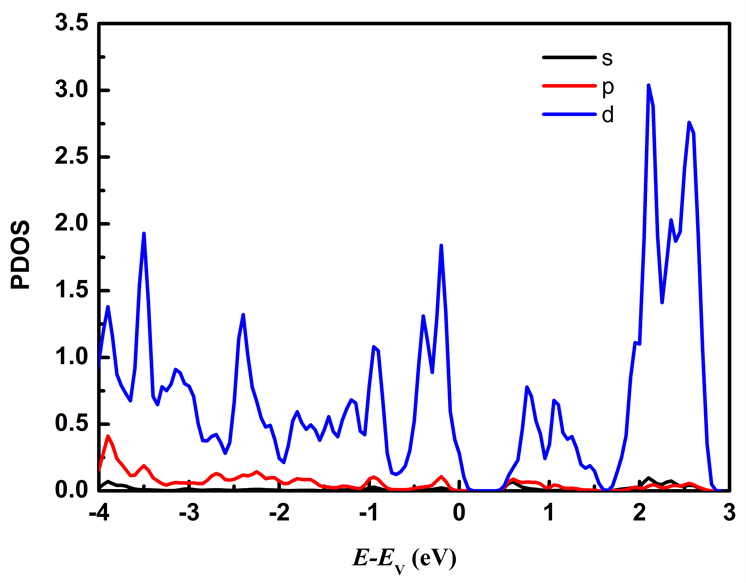
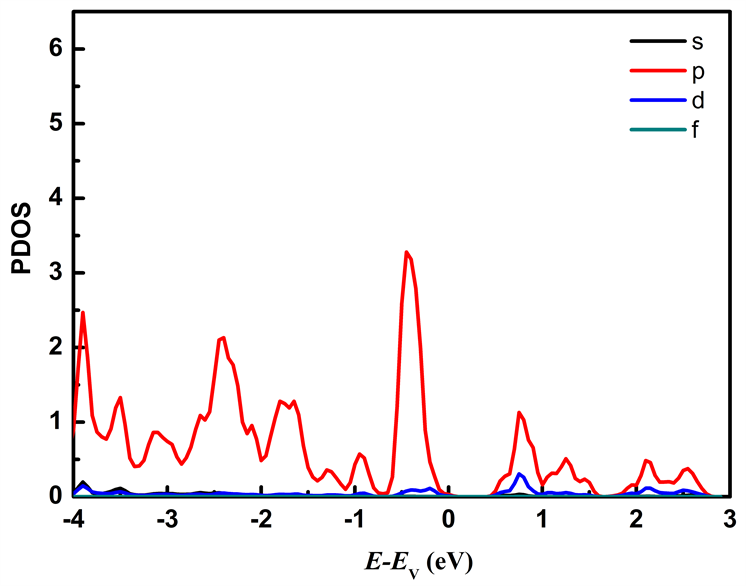
图7分别列出应变为ε = −2%和ε = 2%时Mo原子、W原子和S原子中各电子对态密度的贡献。从图7可以看出在费米能级附近的能态主要由Mo原子的d轨道和W原子的d轨道以及S原子的p轨道所贡献,其中价带顶主要由Mo原子的d轨道、W原子的d轨道以及S原子的p轨道贡献,施加压缩应力时Mo原子的d轨道对价带顶的贡献比施加拉伸应力时的贡献大;导带底主要由Mo原子的d轨道贡献。
 (a) Mo的分波态密度
(a) Mo的分波态密度
 (b) W的分波态密度
(b) W的分波态密度
 (c) S的分波态密度
(c) S的分波态密度
 (d) Mo的分波态密度
(d) Mo的分波态密度
 (e) W的分波态密度
(e) W的分波态密度
 (f) S的分波态密度
(f) S的分波态密度
Figure 7. The partial density of states of MoS2/WS2 nanoribbion under different stress: (a), (b), (c) represent the partial density of states of Mo, W, and S atoms at ε = −2%; (d), (e), (f) represent the partial density of states of Mo, W, and S atoms at ε = 2%
图7. 不同应力下MoS2/WS2纳米带的分波态密度:(a)、(b)、(c)分别为ε = −2%时Mo、W、S原子的分波态密度;(d)、(e)、(f)分别为ε = 2%时的Mo、W、S原子的分波态密度
4. 结论
通过基于密度泛函理论的第一性原理方法,研究了MoS2/WS2超晶格的能带结构和态密度,双轴拉伸应力以及压缩应力对MoS2/WS2超晶格纳米带电子结构的影响。研究发现单层MoS2/WS2超晶格都是直接带隙半导体,其带隙宽度为1.724 eV,且价带顶和导带底位于同一高对称点K点。未施加应力时,MoS2/WS2纳米带为直接带隙,带隙宽度为0.537 eV。对MoS2/WS2纳米带施加双轴拉伸应力后其带隙随着应力的增加而增加,施加压缩应力后带隙宽度随应力的增加而减小,且原子间距以及键长键角的变化影响原子轨道的耦合强度,因而能带结构发生变化。根据MoS2/WS2纳米带的分波态密度分析,得到Mo原子的d轨道对MoS2/WS2纳米带的导带底贡献大,Mo原子的d轨道、W原子的d轨道和S原子的p轨道对MoS2/WS2纳米带的价带顶有贡献。通过对MoS2/WS2超晶格以及纳米带能带结构以及态密度的研究得到能带结构变化的原因,为未来二维过渡金属硫化物应用提供了理论依据。
文章引用
王金华,聂亚婷. MoS2/WS2超晶格电子性质的研究
Study on Electronic Properties of MoS2/WS2 Superlattice[J]. 材料科学, 2023, 13(08): 726-734. https://doi.org/10.12677/MS.2023.138079
参考文献
- 1. Novoselov, K.S., Geim, A.K., Morozov, S.V., Jiang, D., Zhang, Y., Dubonos, S.V., Grigorieva, I.V. and Firsov, A.A.
- 2. (2004) Electric Field Effect in Atomically Thin Carbon Films. Science, 306, 666-669.
- 3. https://doi.org/10.1126/science.1102896
- 4. Gusakova, J., Wang, X., Shiau, L.L., Krivosheeva, A., Shaposhnikov, V., Borisenko, V., Gusakov, V. and Tay, B.K.
- 5. (2017) Electronic Properties of Bulk and Monolayer TMDs: Theoretical Study within DFT Framework (GVJ-2e Method).
- 6. Physica Status Solidi A, 214, Article ID: 1700218. https://doi.org/10.1002/pssa.201700218
- 7. Butler, S.Z., Hollen, S.M., Cao, L., Cui, Y., Gupta, J.A., Gutiérrez, H.R., Heinz, T.F., Hong, S.S., Huang, J., Ismach,
- 8. A.F., et al. (2013) Progress, Challenges, and Opportunities in Two-Dimensional Materials beyond Graphene. ACS Nano,
- 9. 7, 2898-2926. https://doi.org/10.1021/nn400280c
- 10. Radisavljevic, B., Radenovic, A., Brivio, J., Giacometti, V. and Kis, A. (2011) Single-Layer MoS2 Transistors. Nature
- 11. Nanotechnology, 6, 147-150. https://doi.org/10.1038/nnano.2010.279
- 12. Mak, K.F., Lee, C., Hone, J., Shan, J. and Heinz, T.F. (2010) Atomically Thin MoS2: A New Direct-Gap Semiconductor.
- 13. Physical Review Letters, 105, Article ID: 136805. https://doi.org/10.1103/PhysRevLett.105.136805
- 14. Albe, K. and Klein, A. (2002) Density-Functional-Theory Calculations of Electronic Band Structure of Single-Crystal
- 15. and Single Layer WS2. Physical Review B, 66, Article ID: 073413. https://doi.org/10.1103/PhysRevB.66.073413
- 16. Böker, T., Severin, R., Müller, A., Janowitz, C. and Manzke, R. (2001) Band Structure of MoS2, MoSe2, and
- 17. α—MoTe2: Angle-Resolved Photoelectron Spectroscopy and Ab Initio Calculations. Physical Review B, 64, Article ID:
- 18. 235305. https://doi.org/10.1103/PhysRevB.64.235305
- 19. Kim, J.S., Maity, N., Kim, M., et al. (2022) Strain-Modulated Interlayer Charge and Energy Transfers in MoS2/WS2
- 20. Heterobilayer. ACS Applied Materials & Interfaces, 14, 46841-46849. https://doi.org/10.1021/acsami.2c10982
- 21. Farkous, M., Bikerouin, M., Thuan, D.V., Benhouria, Y., et al. (2020) Strain Effects on the Electronic and Optical
- 22. Properties of Van der Waals Heterostructure MoS2/WS2: A First-Principles Study. Physica E: Low-Dimensional Systems
- 23. and Nanostructures, 116, Article ID: 113799. https://doi.org/10.1016/j.physe.2019.113799
- 24. Pieli, B., Novko, D., Raki, I.R., Cai, J., Petrovi, M., Ohmann, R., Vujičić, N., et al. (2021) Electronic Structure of Quasi-
- 25. Freestanding WS2/MoS2 Heterostructures. ACS Applied Materials & Interfaces, 13, 50552-50563.
- 26. https://doi.org/10.1021/acsami.1c15412
- 27. Ramasubramaniam, A., Naveh, D., et al. (2011) Tunable Band Gaps in Bilayer Transition-Metal Dichalcogenides.
- 28. Physical Review B, 84, Article ID: 205325. https://doi.org/10.1103/PhysRevB.84.205325
- 29. Yun, W.S., Han, S.W., et al. (2012) Thickness and Strain Effects on Electronic Structures of Transition Metal Dichalcogenides:
- 30. 2H-MX2 Semiconductors (M = Mo, W; X = S, Se, Te). Physical Review B, 85, Article ID: 033305.
- 31. https://doi.org/10.1103/PhysRevB.85.033305
- 32. Johari, P. and Shenoy, V.B. (2012) Tuning the Electronic Properties of Semiconducting Transition Metal Dichalcogenides
- 33. by Applying Mechanical Strains. ACS Nano, 6, 5449-5456. https://doi.org/10.1021/nn301320r
- 34. Scalise, E., Housa, M., Pourtois, G., Afanas’ev, V. and Stesmans, A. (2012) Strain-Induced Semiconductor to Metal
- 35. Transition in the Two-Dimensional Honeycomb Structure of MoS2. Nano Research, 5, 43-48.
- 36. https://doi.org/10.1007/s12274-011-0183-0
- 37. Kośmider, K. and Fernández-Rossier, J. (2013) Electronic Properties of the MoS2-WS2 Heterojunction. Physical Review
- 38. B, 87, Article ID: 075451. https://doi.org/10.1103/PhysRevB.87.075451
NOTES
*通讯作者。