Advances in Clinical Medicine
Vol.
13
No.
03
(
2023
), Article ID:
63563
,
6
pages
10.12677/ACM.2023.133705
缺血预处理在肾移植缺血再灌注中的研究进展
钟明轩,张克勤*
重庆医科大学附属第二医院泌尿肾病中心,重庆
收稿日期:2023年2月27日;录用日期:2023年3月24日;发布日期:2023年3月31日

摘要
器官获取与移植不可避免面对缺血再灌注损伤,引起细胞内环境改变、炎症反应、活性氧聚集等,影响移植物功能延迟恢复及远期移植器官结局。尽管现阶段有较多研究改善缺血再灌注损伤,使得器官移植1年存活率有所提高,但近年来移植物的长期存活率并没有显著改变。从Murry等人在1986年发现缺血预处理(IPC)对器官移植的潜在益处后,在心脏保护方面取得显著成绩,其他证据表明在肾移植中也有明显效果。缺血预处理通过激活短暂非致死性缺氧环境,体液通讯、神经元刺激、循环免疫细胞的系统修饰和低氧诱导基因的激活保护机体免受更长时间的潜在致死性缺血。本文主要介绍缺血预处理的相关机制。
关键词
缺血预处理(IPC),缺血再灌注损伤(IRI),肾移植

Research Progress of Ischemic Preconditioning with Ischemia Reperfusion Injury of Renal Transplantation
Mingxuan Zhong, Keqin Zhang*
Urinary Nephropathy Center, The Second Affiliated Hospital of Chongqing Medical University, Chongqing
Received: Feb. 27th, 2023; accepted: Mar. 24th, 2023; published: Mar. 31st, 2023

ABSTRACT
Organ acquisition and transplantation inevitably face ischemia-reperfusion injury, causing intracellular environment changes, inflammatory response, reactive oxygen species accumulation, etc., affecting the delayed recovery of graft function and long-term outcomes of transplanted organs. Although there are many studies at present to improve the 1-year survival rate of organ transplantation after ischemia reperfusion injury, the long-term survival rate of grafts has not changed significantly in recent years. Since the discovery by Murry et al. in 1986 of the potential benefits of ischaemic preconditioning (IPC) for organ transplantation with significant results in cardio protection, and other evidence has shown that it also has significant effects in kidney transplantation, ischemic preconditioning protects the organism from potentially lethal ischemia of longer duration by activating a transient nonlethal hypoxic environment, humoral communication, neuronal stimulation, systemic modification of circulating immune cells, and activation of hypoxia inducible genes. This review focuses on the mechanisms involved in ischemic preconditioning.
Keywords:Ischemic Preconditioning, Ischemia Reperfusion Injury, Kidney Transplantation

Copyright © 2023 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/


1. 肾移植中的缺血再灌注损伤
肾脏缺血再灌注损伤(IRI),引起肾功能质量减少、移植血管损伤、慢性缺氧以及随后的纤维化,导致了后来的移植物丢失,降低移植物存活率,组织学上的特征是动脉内膜增厚、肾小球硬化、肾小管间质纤维化和肾小管萎缩。这些组织学变化被认为是与免疫和非免疫因素相关的移植肾累积损伤的最终结果 [1] 。当IRI发生之初,因ATP耗竭,线粒体功能障碍,NA/K泵失活,阳性离子分布异常,细胞内PH值改变、Ca离子潴留,进而引起线粒体膜通透性转换孔(mPTP)开放,向胞质释放凋亡蛋白,细胞色素C等,引起细胞凋亡。再灌注恢复时,产生大量活性氧(ROS),进一步损伤移植物器官 [2] 。移植后,坏死细胞细胞膜破裂,启动DAMPs模式,如HSP、HMGB-1、DNA片段等,从损伤的细胞中释放出来,激发无菌的炎症反应。一旦释放,它们就会连接并激活免疫细胞和实质细胞上表达的模式识别受体(PRR),如Toll样受体(TLRs) TLR2、TLR4。PRR信号通路的激活最终会在受影响的细胞内产生促炎细胞因子和细胞死亡信号,导致一连串的肾损伤 [3] 。有相关研究表明,受损肾小管上皮细胞释放的DAMP也可直接激活健康肾小管上皮细胞的PRR信号,上调促炎症细胞因子 [4] 。固有免疫同样发挥作用,促炎因子及TNF,被受者巨噬细胞 + 树突状细胞识别,直接攻击移植物,而后抗原递呈引起T cell活化,这些T细胞通过细胞毒性CD8 T细胞或促进CD4 T细胞释放细胞因子,进一步损伤移植器官,并逐渐成为慢性排斥的主要病因 [5] 。另外一种现象是发生免疫偏倚,促炎反应活性强于抗炎反应,缺血–再灌注表现出一种特殊类型的免疫反应,这种免疫反应将平衡转移到促炎症的Th1和Th17反应,而不是抗炎的调节T细胞(Treg)反应 [6] 。除细胞免疫途径外,DAMPs模式也同时放大了体液免疫的作用,增加抗体介导的排斥反应 [7] 。在IRI中,缺血缺氧有很强的促纤维生成作用,局部炎症使细胞因子往受损部位迁徙,加重肾小管间质细胞的纤维化反应。肾小管间质纤维化的主要细胞事件包括:炎症细胞的渗透;成纤维细胞的激活和扩张;大量细胞外基质成分的产生和沉积;以及肾小管萎缩和微血管疏松。而后若炎症未能清楚消退,则行成炎症、组织损伤和纤维化的恶性循环 [8] 。微血管障碍也同时起到不可忽视的负面作用,移植肾血管部分出现无血液再通(no-reflow)的现象,血运重新建立后,缺血区并不能得到充分的灌注,许多因素被提出来解释这一现象,如:1) 血管舒缩介质之间的失衡,主要因为炎症介质引起微血管调节障碍,Ach、NO等扩血管物质反应不明显,内皮素、血管紧张素等缩血管物质反应明显。2) 在再灌注过程中,促凝血途径占主导地位,导致肾小管周围和肾小球毛细血管内纤维蛋白沉积和血小板聚集。这些过程可导致微血栓,导致肾血流灌注受损和肾小球滤过率降低。3) 内皮充血,内皮通透性增加导致间质水肿,压迫肾小管周围毛细血管,白细胞黏附增加,以及白细胞在血管外聚集 [9] 。损伤后脱落的细胞及小管内蛋白堵塞于小管内,引起梗阻 [10] 。肾缺血–再灌注损伤仍然是影响供肾长时间保存的主要因素。如何减轻移植肾IRI也日渐成为重要的临床问题之一。缺血预处理技术的开展可能是一个未来的方向。
2. 常见IPC的几种方式
细胞对缺氧/缺血的反应是双峰的。最初有一种适应性保护性条件反应,这种反应在损伤持续时转变为细胞死亡。不同的预处理包括短暂暴露于缺血、轻度热休克和某些药物激活内源性防御机制,以保护细胞或器官免受随后的持续缺血损伤。这就是所谓的缺血耐受。已有证据表明对移植后转归有益,缺血预适应的保护窗在心脏领域已被证明出现在两个“窗口”中,初始保护期在预适应刺激后立即出现并持续1~4小时,延迟或“第二保护窗口”出现在预适应后24小时,并持续24~72小时 [11] ,但肾移植领域还未定论。现在主要通过三种方式:1) 物理IPC,通过在人为干预下暴露于暂短缺血缺氧,手臂或腿部缺血是最常使用的,少见肠系膜前动脉或左肾动脉的预处理刺激,对整个生物体进行IPC;2) 体外细胞IPC,体外将细胞低氧处理,随后注射入体内;3) 生物诱导IPC,通过化学或药理学稳定来模拟缺氧信号HIF-1α传导。我们讨论其可能存在的机制,试图选择更好的方案应对肾移植IRI发生率 [12] 。然而现在主要的证据都来源于动物实验,临床证据偏少。
3. IPC潜在机制
1) HIF-α:肾脏中HIF是一种异源二聚体转录因子,由HIF-α活性亚基和HIF-β结构亚基组成,氧调节的HIF-1α亚单位被O2依赖的脯氨酰羟化酶(PHDs)快速降解,PHDs在正常氧条件下靶向其O2依赖的结构域。相反,在低氧条件下,HIF-1α是稳定的,并与HIF-1β二聚化,激活参与低氧反应的几个基因的转录,如通过红细胞增多症促进氧转运的红细胞生成素(EPO)和促进血管生成的血管内皮生长因子(VEGF) [13] 。但长时间激活HIF,被认为通过与TGF-β1相互作用促进纤维化,一项关于39名肾移植受者发现了这种现象 [14] 。在另外一项实验研究研究肾移植物中缺氧诱导因子HIF-1α通路与不同程度缺血相关性,发现高度缺血性损伤会导致HIF-1α通路的慢性激活,使其偏离血管生成的有益激活 [15] 。轻中度缺血缺氧能发挥HIF-1的正向生理作用,重度则引起损伤,但如何划分缺血程度仍然有争议,暂无定论。
2) mPTP:线粒体膜通透性转换孔(mitochondrial permeability transition pore, mPTP)是调节线粒体膜通透性改变的关键环节,以此稳定和释放凋亡相关因子来调控细胞。ATP耗竭、线粒体钙超载、氧化应激是mPTP开放的主要诱导因素,这可以被CsA抑制。缺血预处理似乎是通过抑制再灌注时的mPTP开放来介导的,进而减少IRI对器官带来的损害。发生缺血时,一系列解偶联氧化磷酸化和引起线粒体肿胀,mPTP开放,导致细胞内间隙中细胞色素C、琥珀酸和线粒体DNA等物质的释放,这些物质能够激活细胞死亡程序,如细胞凋亡或(调节的)坏死,介导细胞死亡 [16] 。腺苷、缓激肽、阿片是IPC的触发因子,可能通过神经激素途径发挥作用,激活G蛋白偶联受体和信号蛋白激酶。这种激活打开KATP通道,导致mPTP抑制。IPC通过PCK的磷酸化,开放ATP依赖的线粒体钾通道(Katp),减少缺血期间线粒体钙超载,并抑制再灌注时的mPTP开放。预防mPTP的开放已被证明是减少IRI的有效策略。
3) NO:NO似乎扮演着双重角色,既可以是预处理的触发剂,也是预处理的介质。最初的NO信号可以作为诱导HIF-1活性的触发器,导致随后的NOS+的表达和更多的NO产生,然后通过iNOS介导保护作用,如增强血管舒张、能量代谢、葡萄糖摄取、血管生成、红细胞生成、细胞活力、增殖和分化。通过HIF-1α的稳定,IPC促进了与维持细胞完整性和细胞代谢有关的多种基因的表达,并通过HO-1防止大量ROS的产生。HIF-1α通过iNOS激活和NO产生直接促进Akt存活途径和NO途径。后者可通过激活KATP通道抑制mPTP开放,并限制ROS过度产生 [12] 。IPC刺激启动eNOS信号,释放的NO导致线粒体KATP通道开放,触发氧化还原信号,引起线粒体膜电位的耗散,进而降低通过线粒体钙单转运体(MCU)摄取钙的动力,防止钙超载。其他提出的保护机制包括通过增加NO释放和增加热休克蛋白表达来抑制炎症反应,减少细胞因子释放来提供细胞保护 [17] 。
4) mir-21:miR-21在缺血再灌注损伤相关过程中的双刃剑效应。miR-21调节的通路同时促进肾脏I/R损伤后的保护/修复或损伤。在I/R的初始阶段,miR-21上调在I/R损伤中提供保护,促进血管生成,抑制炎症反应,减少细胞凋亡,但同时,也协同TGF-1参与肾脏的纤维化过程。在肾脏中,miRs已被证明在肾脏发育和肾脏IRI中起着至关重要的作用,并且在缺氧有益作用的研究中发现了microRNAs调节的证据 [18] 。徐的实验发现,将小鼠肾脏进行15 min肾缺血预处理能显着增加miR-21的表达,并在4天后诱导的缺血再灌注损伤显着减轻。随后敲低miR-21并显着加剧随后的小鼠肾脏缺血再灌注损伤。敲低miR-21导致程序性细胞死亡蛋白4 (miR-21的促凋亡靶基因)显着上调,并显着增加肾小管细胞凋亡。但在没有缺血性预处理的情况下,单独敲低miR-21不会显着影响小鼠肾脏的缺血再灌注损伤 [19] 。这表明miR-21的保护作用可能依赖于IPC对HIF-1的诱导。miR-21的保护机制涉及较多信号通路和分子,比如内皮一氧化氮合酶、热休克转录因子1和HSP70。其中较为明确的是与PTEN-PI3K/Akt信号通路的调节作用有关。据报道,10号染色体(PTEN)上缺失的miR-21靶基因产物是PI3K/Akt信号通路的抑制剂。PI3K/Akt信号通路在缺氧、生长因子和一氧化氮诱导的HIF表达和激活中起重要作用。PTEN已经被证明可以减弱缺氧介导的Akt和HIF-1的激活。稳定和抑制HIF-1调节基因表达。这些结果表明,miR-21可以间接促进缺氧条件下HIF-1的稳定和活性,并与HIF-1一起作用形成正反馈环 [20] 。
5) GSK3β:GSK3β是RISK、SAFE途径的下游靶点,这是一种调节多种细胞过程(包括凋亡、生长和代谢)的蛋白激酶。GSK-3β的磷酸化和失活与IPC保护作用有关。缺血再灌注损伤发生后,氧化还原敏感性GSK3β的活性上调。这将促进NK-kb及Nrf2磷酸化,NK-κB放大促炎基因的转录;Nrf2使得抗氧化基因表达和反应降低。缺血预处理可能通过增强RISK (P13K/AKT)、SAFE (JAK/STAT3)两种信号,来抑制GSK3β,从而保护IRI损伤。降低GSK3β活性也能够稳定线粒体结构,降低线粒体通透性转换孔敏感性,阻碍mPTP通道打开 [21] 。Nrf2活性增加,转位至细胞核,在此诱导HO-1表达,HO-1是一种是一种重要的抗氧化酶,能够抵御过氧化物、ROS等,从而进一步保护器官(见图1)。
6) NF-κB:激活NF-κB是炎症信号通路的中心部分,适度激活可发生保护作用,过度激活则会诱发炎症反应带来损害。HIF-1的升高可激活NF-kb在适度的范围,继而激活Akt-eNOS信号通路,起保护作用 [22] 。有其他研究表明,NF-kb不仅参与炎症,还参与细胞周期活动。IPC后内源性HMGB1的释放通过HMGB1与其受体TLR4的结合导致肾小管上皮细胞短暂的细胞周期停滞。在分子水平上,激活的AMPKα磷酸化p65亚单位诱导NF-κB的激活,并诱导Sema5b的上调引起短暂的保护性G1细胞周期停滞。细胞停滞似乎是一种自我保护程序,使得实验中经历IRI的小鼠展现出较低的AKI发生率,肌酐水平相对较低 [23] 。
7) 阿片、缓激肽、腺苷:腺苷、缓激肽、阿片是IPC的触发因子,所有这三条道路最终都汇聚在PKC。缓激肽与阿片的通路类似,与G蛋白偶联受体结合,吸引P13K,PI3K产生的磷酸化脂质代谢产物磷脂酰肌醇3,4,5-三磷酸和磷脂酰肌醇3,4-二磷酸诱导Akt转位到质膜,P13K/Akt的激活募集eNOs-NO-GC-cGMP-PKG途径,并通过该级联反应打开Katp通道,减少Ca离子超载,从而抑制mPTP开放 [24] 。腺苷信号似乎绕过了Katp通道,与其受体结合后直接水解含膜肌醇磷脂,产生DAG来刺激PKC的转移和激活 [25] 。大致过程见图1。
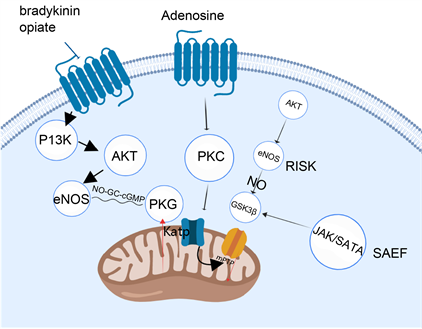
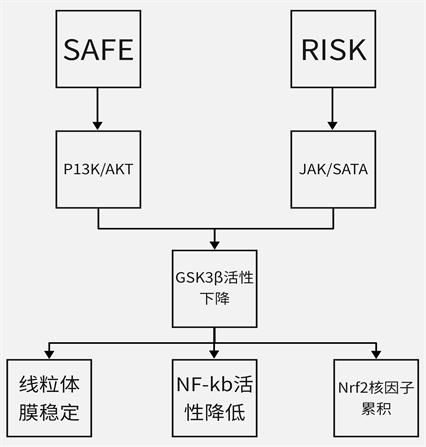
Figure 1. The mechanisms of opioid, bradykinin, adenosine and RISK and SAFE pathway
图1. 阿片、缓激肽、腺苷及RISK、SAFE的机制
4. 总结
目前,IPC对IRI保护作用的研究大多仍停留在动物实验阶段,IPC从实验领域转化为临床实践仍然面临着巨大的挑战及困难。但IPC因其安全性及方便性值得人们关注,不清楚的地方还有很多,但随着更多机制的探索以及临床实验的尝试,有望取得进步和突破。希望此文能给带来一些启示。
文章引用
钟明轩,张克勤. 缺血预处理在肾移植缺血再灌注中的研究进展
Research Progress of Ischemic Preconditioning with Ischemia Reperfusion Injury of Renal Transplantation[J]. 临床医学进展, 2023, 13(03): 4942-4947. https://doi.org/10.12677/ACM.2023.133705
参考文献
- 1. Zhao, H., et al. (2018) Ischemia-Reperfusion Injury Reduces Long Term Renal Graft Survival: Mechanism and Beyond. EBioMedicine, 28, 31-42. https://doi.org/10.1016/j.ebiom.2018.01.025
- 2. Yan, H.F., et al. (2020) The Patholog-ical Role of Ferroptosis in Ischemia/Reperfusion-Related Injury. Zoological Research, 41, 220-230. https://doi.org/10.24272/j.issn.2095-8137.2020.042
- 3. Shelke, V., et al. (2022) Epigenetic Regulation of Toll-Like Receptors 2 and 4 in Kidney Disease. Journal of Molecular Medicine (Berlin), 100, 1017-1026. https://doi.org/10.1007/s00109-022-02218-y
- 4. DeWolf, S.E., et al. (2022) DAMPs Released from Injured Re-nal Tubular Epithelial Cells Activate Innate Immune Signals in Healthy Renal Tubular Epithelial Cells. Transplantation, 106, 1589-1599. https://doi.org/10.1097/TP.0000000000004038
- 5. Li, L. and Okusa, M.D. (2010) Macrophages, Dendritic Cells, and Kidney Ischemia-Reperfusion Injury. Seminars in Nephrology, 30, 268-277. https://doi.org/10.1016/j.semnephrol.2010.03.005
- 6. Barbi, J., Pardoll, D. and Pan, F. (2013) Metabolic Control of the Treg/Th17 Axis. Immunological Reviews, 252, 52-77. https://doi.org/10.1111/imr.12029
- 7. Fuquay, R., et al. (2013) Renal Ischemia-Reperfusion Injury Amplifies the Humoral Immune Response. Journal of the American Society of Nephrology, 24, 1063-1072. https://doi.org/10.1681/ASN.2012060560
- 8. Liu, Y. (2011) Cellular and Molecular Mechanisms of Renal Fibro-sis. Nature Reviews Nephrology, 7, 684-696. https://doi.org/10.1038/nrneph.2011.149
- 9. Legrand, M., et al. (2008) Renal Hypoxia and Dysoxia after Reper-fusion of the Ischemic Kidney. Molecular Medicine, 14, 502-516. https://doi.org/10.2119/2008-00006.Legrand
- 10. Veighey, K. and MacAllister, R. (2015) Clinical Applications of Remote Ischaemic Preconditioning in Native and Transplant Acute Kidney Injury. Pediatric Nephrology, 30, 1749-1759. https://doi.org/10.1007/s00467-014-2965-6
- 11. Yellon, D.M. and Downey, J.M. (2003) Preconditioning the My-ocardium: From Cellular Physiology to Clinical Cardiology. Physiological Reviews, 83, 1113-1151. https://doi.org/10.1152/physrev.00009.2003
- 12. Bruzzese, L., et al. (2021) Hypoxic Preconditioning in Renal Is-chaemia-Reperfusion Injury: A Review in Pre-Clinical Models. Clinical Science (London), 135, 2607-2618. https://doi.org/10.1042/CS20210615
- 13. Guieu, R., et al. (2020) Adenosine and the Cardiovascular System: The Good and the Bad. Journal of Clinical Medicine, 9, 1366. https://doi.org/10.3390/jcm9051366
- 14. Kellenberger, T., et al. (2015) Expression of Hypoxia-Inducible Factor-1α and Hepatocyte Growth Factor in Development of Fibrosis in the Transplanted Kidney. Transplant International, 28, 180-190. https://doi.org/10.1111/tri.12475
- 15. Delpech, P.O., et al. (2014) Effects of Warm Ischaemia Combined with Cold Preservation on the Hypoxia-Inducible Factor 1α Pathway in an Experimental Renal Autotransplantation Model. British Journal of Surgery, 101, 1739-1750. https://doi.org/10.1002/bjs.9611
- 16. Kharechkina, E.S., et al. (2021) Pioglitazone Is a Mild Carrier-Dependent Uncoupler of Oxidative Phosphorylation and a Modulator of Mitochondrial Permeability Transition. Pharmaceuticals (Basel), 14, 1045. https://doi.org/10.3390/ph14101045
- 17. Randhawa, P.K., Bali, A. and Jaggi, A.S. (2015) RIPC for Multiorgan Salvage in Clinical Settings: Evolution of Concept, Evidences and Mechanisms. European Journal of Pharmacology, 746, 317-332. https://doi.org/10.1016/j.ejphar.2014.08.016
- 18. Ho, J., et al. (2008) Podocyte-Specific Loss of Functional mi-croRNAs Leads to Rapid Glomerular and Tubular Injury. Journal of the American Society of Nephrology, 19, 2069-2075. https://doi.org/10.1681/ASN.2008020162
- 19. Xu, X., et al. (2012) Delayed Ischemic Preconditioning Contributes to Renal Protection by Upregulation of miR-21. Kidney International, 82, 1167-1175. https://doi.org/10.1038/ki.2012.241
- 20. Ghafouri-Fard, S., et al. (2021) Regulatory Role of microRNAs on PTEN Signaling. Biomedicine & Pharmacotherapy, 133, Article ID: 110986. https://doi.org/10.1016/j.biopha.2020.110986
- 21. Liu, Z. and Gong, R. (2015) Remote Ischemic Preconditioning for Kidney Protection: GSK3β-Centric Insights into the Mechanism of Action. American Journal of Kidney Diseases, 66, 846-856. https://doi.org/10.1053/j.ajkd.2015.06.026
- 22. Crowley, L.E. and McIntyre, C.W. (2013) Remote Is-chaemic Conditioning-Therapeutic Opportunities in Renal Medicine. Nature Reviews Nephrology, 9, 739-746. https://doi.org/10.1038/nrneph.2013.226
- 23. Rossaint, J., et al. (2022) Remote Ischemic Preconditioning Causes Transient Cell Cycle Arrest and Renal Protection by a NF-κB-Dependent Sema5B Pathway. JCI Insight, 7, e158523. https://doi.org/10.1172/jci.insight.158523
- 24. Ong, S.B., et al. (2015) Role of the MPTP in Conditioning the Heart-Translatability and Mechanism. British Journal of Pharmacology, 172, 2074-2084. https://doi.org/10.1111/bph.13013
- 25. Cohen, M.V., et al. (2001) Acetylcholine, Bradykinin, Opioids, and Phe-nylephrine, but Not Adenosine, Trigger Preconditioning by Generating Free Radicals and Opening Mitochondrial K(ATP) Channels. Circulation Research, 89, 273-278. https://doi.org/10.1161/hh1501.094266
NOTES
*通讯作者。