Advances in Microbiology
Vol.
08
No.
02
(
2019
), Article ID:
30710
,
9
pages
10.12677/AMB.2019.82010
Analysis of Bacterial Communities in Soybean Rhizosphere Based on High Throughput Sequencing
Lin Wang1*#, Lulu Li2, Ruirui Liu2, Xin Liu2
1Department of Medical Technology, Xi’an Medical University, Xi’an Shaanxi
2Key Laboratory of Resource Biology and Biotechnology in Western China, Ministry of Education, Northwest University, Xi’an Shaanxi

Received: May 22nd, 2019; accepted: Jun. 4th, 2019; published: Jun. 11th, 2019

ABSTRACT
The changes of microorganisms community soil have important effects on soybean plants, so it is necessary to study the changes. The rhizosphere soil of different growth stages was studied, and the bacteria were sequenced using the Illumina HiSeq 2500 sequencing platform in the rhizosphere soil. After genomic DNA extraction, PCR product amplification and purification, construction of the library and sequencing data analysis, the species annotation of the high throughput sequencing experiment showed that the relative abundance of Proteobacteria, Acidophilus, and Actinomycetes was the highest in the samples. Alpha diversity experiment showed that the diversity is more abundant at maturity.
Keywords:Soybean, Rhizosphere Soil, High-Throughput Sequencing, Bacterial Community
基于高通量测序的大豆根际土壤细菌群落的 研究
王琳1*#,李璐璐2,刘瑞瑞2,刘欣2
1西安医学院医学技术系,陕西 西安
2西北大学,西部生物资源与现代生物技术教育部重点实验室,陕西 西安
收稿日期:2019年5月22日;录用日期:2019年6月4日;发布日期:2019年6月11日

摘 要
大豆根际土壤中微生物群落的变化对大豆植株会产生一定影响,所以研究其变化很有必要。以不同生长阶段的大豆根际土壤为研究对象,利用Illumina HiSeq 2500测序平台对根际土壤中细菌进行测序。经过基因组DNA提取、PCR产物扩增纯化、构建文库以及测序数据分析,物种注释结果表明样品中变形菌门、酸杆菌门、和放线菌门的相对丰度最高。Alpha多样性结果表明成熟期的多样性更加丰富。
关键词 :大豆,根际土壤,高通量测序,细菌群落

Copyright © 2019 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 引言
当今世界土地上种植的大豆具有资源丰富、高价值、多样性等特点,所以大豆成为种植最广泛的豆类植物之一,以至于在农副产品中占有很重要的位置 [1]。如今,中国对大豆的需求正在增加,但主要依赖于进口 [2]。所以,提高大豆产量是我国较为迫切的问题。中国在农业生产过程中仍然延续了前人的经验,如农药、化肥等的使用。虽然使用化肥可以在一定时间内提高农作物产量,但是长时间使用会使得土壤很难恢复到以往的状态,此外,过量使用会导致土壤酸化、土壤肥力下降和水体富营养化等问题,进而影响到我国农业的发展 [3]。而近年来,越来越多的微生物肥料已崭露头角,在改善土壤环境方面微生物肥料比传统的方法更加有优势 [4]。
1904年,德国的Hiltner提出了“根际”概念来描述植物根系分泌物对土壤微生物的影响 [5]。这种效应主要是根际分泌物对其周围根际微生物的群落结构和生态分布的根际效应 [6]。我们现在面临的一个问题是探索自然环境中微生物群落的结构、功能和活动。为了解决这个问题,近些年来,许多科学家已经探索出一些技术方法。目前,最广泛使用的是高通量测序,其测序平台包括 Illumina (如HiSeq,MiSeq),Roche454 GS FLX,SOLiD5500系列以及Ion Torrent/ Ion Proton平台。其中Illumina系统数据量大、成本相对较低和SOLiD的系统准确度高的优势让其成为现在二代测序研究的主要手段 [7]。
本文利用大豆在幼苗期和成熟期两个不同生长阶段的根际土壤作为实验材料,然后运用Illumina测序平台对其根际土壤中细菌的16SrRNA进行高通量测序,将得到的数据进行分析并比较这两个时期的细菌群落结构的不同,这一研究为以后豆科植物不同阶段土壤微生物的细菌群落结构的变化提供理论基础。
2. 材料与方法
2.1. 材料
2.1.1. 研究区概况
本次研究对象为大豆根际土壤,所用土壤样本采集于陕西省咸阳市白良寨村(位于陕西省咸阳市秦都区双照街道东南方向,距离咸阳市区10公里;34˚11'N,107˚38'E,暖温带大陆性季风气候,年平均气温 13.4℃,降水量519.2 mm,日照2175 h)大豆田。
2.1.2. 样品采集
2016年6月24日和2016年9月30日采用“S形采样法”在大豆田中选择5个采样点,使用抖根法 [8] 对根际土壤进行取样:首先从土壤中将植物的根部轻轻挖出,然后轻轻抖落和根部相结合的部分土壤,再用无菌小刷子刷取,将取得的土壤样品混合到一起,最后通过2 mm筛。最后将其均匀分成三等份放置于−20℃冰箱保存。将2016年6月24日(苗期)采集的样品编号DWB;将2016年9月30日(成熟期)采集的样品编号为DYB。
2.2. 方法
2.2.1. 土壤总DNA的提取与电泳检测
土壤总DNA的提取:通过美国MoBio的PowerSoil DNA Isolation Kit试剂盒,并将提取的总DNA进行琼脂糖凝胶电泳和核酸定量分光光度计(NanoDrop,美国)检测 [9] ,检测合格以后,密封送往北京百迈克公司进行测序。
2.2.2. 试验上机流程
整个试验流程包括对土壤样品中细菌16SrRNA进行PCR扩增,选择细菌通用的引物338F (5'-ACTCCTACGGGAGGCAGCA-3')和806R (5'-GGACTACHVGGGTWTCTAAT-3')对细菌的16SrDNA进行扩增。扩增条件为如表1所示,PCR扩增过程:预变性(95℃) 5 min,变性(95℃) 1 min,退火(50℃) 1 min,延伸(72℃) 1 min,最终延伸(72℃) 7 min。然后进行产物的纯化,建立文库,进行质量检查,再使用Illumina HiSeq 2500平台检查质量并测序 [10]。以上测序文库的构建、质检和上机测序流程由北京百迈克有限公司提供。高通量测序因其特有的优势,比如测序深度较高,成本低,数据量较大以及信息量较丰富已经在现代土壤微生物的研究中越来越普遍。

Table 1. PCR reaction system
表1. PCR反应体系
2.2.3. 测序数据分析方法
1) 使用FLASH V1.2.7软件,得到Raw Tags。2) 使用Trimmomatic v 0.33软件,得到Clean Tags。3) 使用Uchime V4软件鉴定并去除嵌合体序列,以获得有效数据(Effective Tags)。4) Uclust用于QIIME (版本1.8.0)软件中,使用Uclust以97%的相似性水平聚类序列,并获得样品的OUT分类数据。5) 根据测序结构对门水平和属水平上的细菌菌群丰度进行分析。6) 根据测序得出的结果进行Alpha多样性分析,其中Shannon指数和Simpson指数与细菌群落的多样性有关,Simpson越小,Shannon越大,则细菌群落物种多样性越高。Ace指数、Chao1指数越大,样本丰富度越高。7) Bata多样性分析,用于比较样本之间的差异。
3. 结果与分析
3.1. 样品测序结果分析
将高通量得到的数据进行数据预处理 [11] ,得到的测序长度集中在360~460 bp之间,6个样品产生总共1544个OTU,各样本统计结果见图1。从样品中随机选择一定数量的序列,并计算由这些序列表示的物种数,并通过序列数和物种数绘制稀释曲线,如图2所示。随着测序次数的增加,曲线逐渐变平,表明测序深度足够。

Figure 1. Distribution of OTU numbers in each sample
图1. 各样本OTU个数分布图
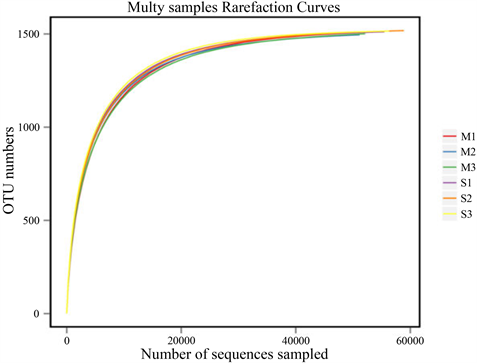
Figure 2. Dilution curve of bacteria sequencing in soybean rhizosphere soil samples
图2. 大豆根际土壤样品细菌测序的稀释曲线
3.2. 土壤细菌结构群落分析
在97%的相似度水平下,利用Mothur软件对OTU数进行分析,然后再与Silva数据库进行相似性比对。结果显示,样品中的细菌分属于25个门,78个纲,123个目,205个科,317个属,234个种。
3.2.1. 门水平群落结构
由图3所知,在两个时期中,苗期(DWB)门水平上按相对丰度的高低排序,优势菌门(丰度 > 4%)是放线菌门(Actinobacteria, 29.76%),酸杆菌门(Acidobacteria, 19.90%),变形菌门(Proteobacteria, 26.38%),绿弯菌门(Chloroflexi, 4.72%),芽单胞菌门(Gemmatimonadetes, 9.22%),拟杆菌门(Bacteroidetes, 5.61%)。成熟期(DYB)的优势菌门(>4%)和成熟期相同,但是相比成熟期来说只有放线菌门(Actinobacteria, 40.36%)的相对丰度增加了,酸杆菌门(Acidobacteria, 17.89%),变形菌门(Proteobacteria, 18.58%),绿弯菌门(Chloroflexi, 4.38%),芽单胞菌门(Gemmatimonadetes, 8.71%),拟杆菌门(Bacteroidetes, 4.74%)的相对丰度都下降了。
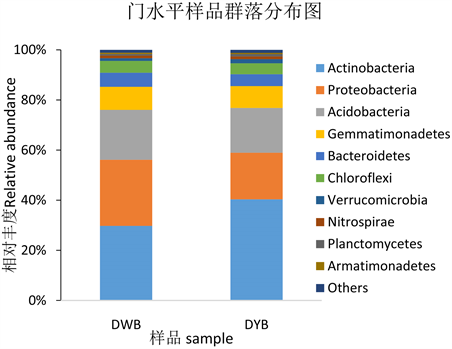
Figure 3. Phylum distribution of phylum level
图3. 门水平物种分布图
3.2.2. 属水平群落结构
由图4所知,属水平上,按相对丰度排序,苗期(DWB)的优势类群(丰度 > 4%)是:bacterium (45.12%),Pseudonocardia (12.02%),Sphingomonas (6.26%),成熟期(DYB)的优势类群(丰度 > 4%)是:bacterium (43.40%),Pseudonocardia (10.02%),RB41 (4.20%)。由此可知不同时期的土壤样品在属水平上的群落结构有所差异。
3.2.3. 门水平和属水平上ANOVA分析
按相对丰度高低排序,由表2、表3可知,在门级水平的前十个门中,除放线菌、芽单胞菌门、硝化螺旋菌门,其他细菌之间皆存在有显著差异,当中属酸菌门的差异较为明显(p = 0.008155);在属水平上排名前十的属中,有显著差异的属为Pseudonocardia (p = 0.000762),Sphingomonas (p = 0.000011),RB41 (p = 0.000481),Bryobacter (p = 0.009852) Pseudarthrobacter (p = 0.000003),Streptonmyces (p = 0.0001),其中差异最为显著的属为Pseudonocardia。

Figure 4. Genus distribution of phylum level
图4. 属水平物种分布图
Table 2. ANOVA variance analysis on relative abundances of major bacterial flora at phylum level
表2. 门水平主要菌群相对丰度ANOVA方差分析
Table 3. ANOVA variance analysis on relative abundances of major bacterial flora at the genus level
表3. 属水平主要菌群相对丰度ANOVA方差分析
3.3. 细菌群落多样性分析
从样品的Alpha多样性指数可以看出细菌群落的丰度和多样性,衡量指数Shannon指数、Simpson指数、Ace指数、Chao1指数以及覆盖率如表4所示。结果显示大豆在成熟期的物种多样性和丰富度比苗期高。
Table 4. Diversity alpha index
表4. Alpha多样性指数统计
注:Sample ID为样品名称;Chao1,Ace,Shannon,Simpson分别表示各个指数;Coverage是样本文库的覆盖率。
3.4. Bata多样性分析
Nonmetric Multidimensional Scaling是一种非线性多维尺度分析,通常用于比较样本之间的差异。所有样品的NMDS图谱如图5所示,图5表明两个样本之间的相似性较低,即生长阶段不同的大豆根际微生物群落结构不同。
4. 讨论与结论
有许多种土壤微生物,它们是栖息在土壤生态系统中的大量生物群落 [12]。土壤中的细菌群落是其重要成分之一,而其细菌群落结构的多样性是和土壤的质量和稳定性息息相关的 [13]。因此,研究土壤微生物是必要的。研究表明,通过以前的传统方法测量的微生物数量很少,约占微生物总数的0.1%至10% [14]。这些传统方法只能检测门、类等水平的微生物类型,因此土壤中的土壤微生物群落结构不能作为一个整体来描述。Illumina的高通量测序技术快速、方便、高度准确。利用高通量测序技术进行土壤微生物研究,大大提高了研究人员对土壤微生物多样性的认识 [15]。已经探索了许多未知的土壤微生物及其在土壤中的功能作用。在本研究中,我们分析了大豆两个不同生长阶段根际土壤中细菌的群落结构和多样性。通过该方法获得的结果表明每个样品的覆盖率高于99%。从另一方面也表明本次的测序结果是能够准确反映样本的情况的 [16]。
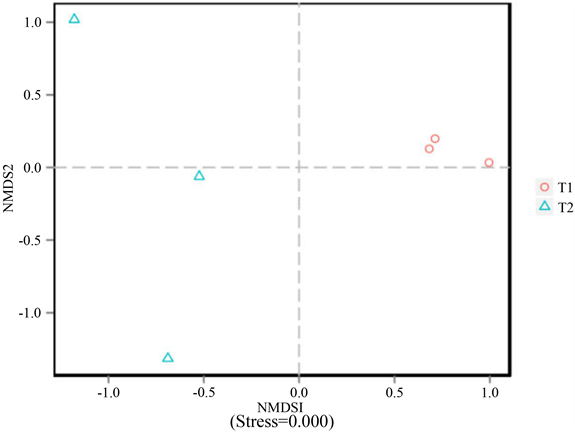
Figure 5. Non-metric multidimensional scaling analysis
图5. NMDS分析
该研究发现,所有样本都含有25个细菌门,其中变形菌、酸杆菌、和放线菌的相对丰度最高。Alpha多样性的研究结果表明,Chao1、Ace和Shannon指数显示成熟期的指数要高于苗期的,所以得出大豆在成熟阶段的细菌群落多样性和丰度要比苗期高。NMDS用于比较样本的组内或组间差异。在本研究中,Stress = 0.0000,表明NMDS分析非常可靠。大量研究证实,根际微生物的数量和群落结构将随着生长期而变化。结果表明,两组样品清晰地位于不同的象限,表明两个样品之间的相似性不高,即生长期不同根际细菌的群落结构不同。大量研究证实,根际微生物的数量和群落结构将随着生长期而变化。
高通量技术可以详尽地反映出不同生长阶段大豆根际微生物群落结构多样性的变化,本研究以不同生长阶段的大豆根际土壤为研究对象,通过微生物菌群组成发现在前后两个不同生长阶段的样品中,门水平和属水平中的优势菌群都发生了变化,而成熟期的菌群比苗期的菌群多样性更加丰富。但是由于高通量技术的局限性,土壤样品中还有未能检测出来的细菌种类,所以仍需进一步探讨研究。
基金项目
国家自然科学基金(41571243),西安医学院配套基金项目(2017PT14);西安医学院国家基金培育项目(2017GJFY16)。
文章引用
王 琳,李璐璐,刘瑞瑞,刘 欣. 基于高通量测序的大豆根际土壤细菌群落的研究
Analysis of Bacterial Communities in Soybean Rhizosphere Based on High Throughput Sequencing[J]. 微生物前沿, 2019, 08(02): 80-88. https://doi.org/10.12677/AMB.2019.82010
参考文献
- 1. Herridge, D.F., Peoples, M.B. and Boddey, R.M. (2008) Global Inputs of Biological Nitrogen Fixation in Agricultural Systems. Plant and Soil, 311, 1-18. https://doi.org/10.1007/s11104-008-9668-3
- 2. 张振华, 刘志民. 我国大豆供需现状与未来十年预测分析[J]. 大豆科技, 2009(4): 16-21.
- 3. 熊峰. 四川大豆根瘤菌遗传多样性和系统发育研究[D]: [硕士学位论文]. 雅安: 四川农业大学, 2014.
- 4. 沈耀耀. 西北部分地区野大豆根瘤菌遗传多样性及系统发育的研究[D]: [硕士学位论文]. 杨凌: 西北农林科技大学, 2011.
- 5. Hiltner, H.A., Rothballer, M., Schmid, M. and Lorenz, H. (2008) A Pioneer in Rhizosphere Microbial Ecology and Soil Bacteriology Research. Plant and Soil, 312, 7-14. https://doi.org/10.1007/s11104-007-9514-z
- 6. 朱丽霞, 章家恩, 刘文高. 根系分泌物与根际微生物相互作用研究综述[J]. 生态环境, 2003, 12(1): 102-105.
- 7. Metzker, M.L. (2010) Sequencing Technologies—The Next Generation. Nature Reviews Genetics, 11, 31-46. https://doi.org/10.1038/nrg2626
- 8. 张福锁, 等. 环境胁迫与植物根际营养[M]. 北京: 中国农业出版社, 1998.
- 9. 秦杰. 长期施肥对黑土肥力及细菌和古菌群落的影响[D]: [硕士学位论文]. 泰安: 山东农业大学, 2015.
- 10. 陈究成. 浙江省地方猪群体遗传结构研究[D]: [博士学位论文]. 杭州: 浙江大学, 2016.
- 11. 王伏伟, 王晓波, 李金才, 等. 施肥及秸秆还田对砂姜黑土细菌群落的影响[J]. 中国生态农业学报, 2015, 23(10): 1302-1311.
- 12. 周群英, 高廷耀. 环境工程微生物学[M]. 北京: 高等教育出版社, 2000.
- 13. 王光华, 金剑, 徐美娜. 植物、土壤及土壤管理对土壤微生物群落结构的影响[J]. 生态学杂志, 2006, 25(5): 550-556.
- 14. Felber, R., Leifeld, J., Horák, J. and Neftel, A. (2014) Nitrous Oxide Emission Reduction with Greenwaste Biochar: Comparison of Laboratory and Field Experiments. European Journal of Soil Science, 65, 128-138. https://doi.org/10.1111/ejss.12093
- 15. Wilson, M.C. and Piel, J. (2013) Metagenomic Approaches for Exploiting Uncultivated Bacteria as a Resource for Novel Biosynthetic Enzymology. Chemistry & Biology, 20, 636-647. https://doi.org/10.1016/j.chembiol.2013.04.011
- 16. 王朝江, 王世清. 基于高通量测序技术对三种太岁样品细菌组成的分析[J]. 湖北农业科学, 2017, 56(13): 2543-2547.
NOTES
*第一作者。
#通讯作者。