Hans Journal of Chemical Engineering and Technology
Vol.
10
No.
05
(
2020
), Article ID:
37509
,
5
pages
10.12677/HJCET.2020.105046
双醋瑞因杂质的合成
代泽琴1,张毅1,2,冯广卫1*
1贵州医科大学药学院,贵州 贵阳
2贵州省化学合成药物研发利用工程技术研究中心,贵州 贵阳

收稿日期:2020年8月17日;录用日期:2020年8月31日;发布日期:2020年9月8日

摘要
本文以芦荟大黄素为起始原料,经氧化、乙酰化、保护、氢解四步反应合成了双醋瑞因杂质;该杂质的合成对仿制药的申报和质量研究有一定意义。其化合物经ESI-MS、IR、1H NMR、13C NMR元素分析确证。
关键词
杂质,分离纯化,合成

Synthesis of Diacerein Impurities
Zeqin Dai1, Yi Zhang1,2, Guangwei Feng1*
1School of Pharmacy, Guizhou Medical University, Guiyang Guizhou
2Guizhou Provincial Engineering Technology Research Center for Chemical Drug R&D, Guiyang Guizhou
Received: Aug. 17th, 2020; accepted: Aug. 31st, 2020; published: Sep. 8th, 2020

ABSTRACT
Using aloe emodin as the starting material, the diacerein impurities were synthesized by oxidation, acetylation, protection and hydrolyzation. The synthesis of the impurity is of certain significance to the application and quality research of generic drugs. The compounds were characterized by ESI-MS, IR, 1H NMR, and 13C NMR.
Keywords:Impurity, Separation and Purification, Synthesis

Copyright © 2020 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/


1. 引言
双醋瑞因 [1] 可通过抑制成骨性细胞RANKL的表达或抑制其上游调控因子而抑制由L-1B刺激的骨吸收,从根本上治疗退行性关节炎等疾病。同时可诱导软骨生成,具有止痛、抗炎及退热作用,不抑制前列腺素合成,对骨关节炎有延缓疾病进程的作用 [2]。有文献报道双醋瑞因的相关合成路径 [3] - [8],但是没有两个已知单乙酰化(4-乙酰基-5-羟基-9,10-二氧-9,10-二氢蒽-2-羧酸(1a);5-乙酰基-4-羟基-9,10-二氧-9,10-二氢蒽-2-羧酸(1b))杂质的合成报道,有关报道也只是采用制备液相分离鉴别 [9] [10],也没有很好地得到产物,即使采用制备液相来分离纯化,成本高昂;并且该化合物不稳定,极易水解。在仿制药的申报及生产过程中需要相关杂质作为对照品 [11],然而国内目前的销售价格在100 mg/2万元左右,因此两个已知杂质会给双醋瑞因化学仿制药的申报带来高昂的成本,研究一条能制备出该杂质的合成工艺很有意义。
基于(1a)、(1b)化合物是一个平面结构,溶解性很差 [12],很难直接通过层析硅胶柱分离纯化,因此我们从改变功能团改变其理化性质的基本原理出发,用溴苄增强羟基和羧基的酯溶性,硅胶柱层析分离纯化,再使用10%的Pd/C催化氢解。本研究以芦荟大黄素为起始原料,经氧化、乙酰化、溴苄保护、钯炭催化还原4步反应,得到双醋瑞因的两个关键杂质(1a)、(1b),合成路径见图1。经ESI-MS,IR、1H NMR、13C NMR、元素分析确证。
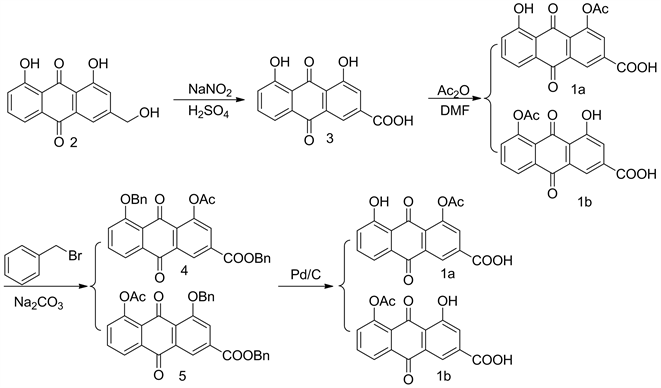
Figure 1. The synthetic route of the title compound
图1. 合成路线图
2. 实验部分
2.1. 主要仪器与试剂
Varian Mercury-400核磁共振仪(美国Varian公司);Vario EL元素分析仪(德国Elementar元素分析系统公司);Magna FT-IR-750光谱仪(美国Nicolet公司);1100型高效液相色谱仪(配有二级管阵列(DAD)检测器,美国Agilent公司);Xevo G2-XS QTof高分辨质谱仪(美国Waters公司);所用试剂均为分析纯。
2.2. 合成
2.2.1. 大黄酸(3)的合成
在250 mL三口烧瓶中加入硫酸120 mL,缓慢加入亚硝酸钠25.55 g (370.40 mmo1),加完升温至120℃后并保温于120℃缓慢加入10.0 g (37.04 mmo1)芦荟大黄素2,加完保温120℃继续搅拌反应4小时;降至室温,将反应液缓慢倒入搅拌的1000 mL冰水中,继续搅拌0.5小时,抽滤,滤饼用水2 × 100 mL洗涤,抽至无液体流出,滤饼105℃干燥,得粉状固体10.0 g,加入N,N-二甲基甲酰胺100 mL加热溶解澄清后,室温放置析晶12小时,抽滤,滤饼用水2 × 40 mL洗,抽至无液体流出,滤饼105℃干燥,得黄色粉末状结晶9.6 g,收率:91.25%。mp 316℃~318℃,文献 [3] (317℃~319℃),1H NMR (400 MHZ, DMSO-d6),δ = 7.42 (dd, J = 1.2 Hz, 8.4 Hz, 1H, Ar-H),7.75~7.77 (m, 2H, Ar-H),7.84 (dd, J = 7.5 Hz, 8.3 Hz, 1H, Ar-H),8.16 (d, J = 1.2 Hz, 1H, Ar-H),11.93 (s, 2H, OH),13.70 (s, 1H, COOH);IRν:3061,2604,1692,1629,1608,1570,1452,1266,1188,1075;ESI-MS m/z:Calcd for C15H8O6Na {[M-H]−}284.032 1,found 284.0323。
2.2.2. 单乙酰化混合物(1a, 1b)的合成
于500 mL圆底瓶中加入9 g (31.69 mmol)化合物3,乙酸酐7 mL (74.05 mmol),1,4-二氧六环90 mL,加完油浴回流反应20小时,[TLC检测(展开剂:V (石油醚):V (乙酸乙酯):V (乙酸) = 20:10:1]。降至60℃减压蒸出大量1,4-二氧六环,加入200 mL水搅拌0.5小时,抽滤,滤饼用50 mL水淋洗一次,抽至无液体流出,滤饼50℃减压蒸干得黄色粉状固体9.5 g,该混合固体未经纯化直接往下合成。
2.2.3. 4-乙酰基-5-苄氧基-9,10-二氧-9,10-二氢蒽-2-羧酸苄酯(4)与5-乙酰基-4-苄氧基-9,10-二氧-9,10-二氢蒽-2-羧酸羧酸苄酯(5)的合成
将混合固体9 g加入到250 mL圆底瓶中,再加入N,N-二甲基乙酰胺90 mL,无水碳酸钠5.8 g,溴化苄4.18 mL,加完升温至100℃搅拌反应4小时,[TLC检测(展开剂:V (石油醚):V (乙酸乙酯) = 2:1]几乎无混合物(1a, 1b)点。停止加温,降至室温加入200 mL CH2Cl2,冰水400 mL,加完搅拌15 min,分出CH2Cl2,CH2Cl2层2 × 200 mL水洗,CH2Cl2无水硫酸钠干燥,40℃减压回收CH2Cl2得浸膏状产物,加入100 mL石油醚有红棕色固体析出,搅拌1小时,抽滤,滤饼25 mL石油醚淋洗一次抽干,滤饼45 ℃减压干燥,得红棕色混合粉状固体11 g。
将11 g红棕色粉状固体溶于110 mL乙酸乙酯中,加入22 g硅胶拌样干法上样过硅胶层析柱,[洗脱液,V (石油醚):V (乙酸乙酯) = 3:1)]洗脱液分别减压蒸干得化合物4 2.6 g,ESI-MS m/z:Calcd for C31H22O7Na {[M-H]−} 505.1366,found 505.1367;化合物5 1.2 g,ESI-MS m/z:Calcd for C31H22O7Na {[M-H]−} 505.1366,found 505.1369。
2.2.4. 4-乙酰基-5-羟基-9,10-二氧-9,10-二氢蒽-2-羧酸 (1a)的合成
将2.5 g化合物4加入到50 mL圆底瓶中,加入THF 25 mL,10%的Pd/C 0.25 g,加完氢气真空切换3次,后通氢气室温搅拌反应12小时,[TLC检测(展开剂:V (石油醚):V (乙酸乙酯):V (乙酸) = 20:10:1]。抽滤,滤液40℃减压蒸干得浅红棕色固体1.6 g,加入乙酸乙酯16 mL搅拌1小时后抽滤,滤饼5 mL乙酸乙酯淋洗一次,抽干,滤饼45℃减压干燥,得浅红棕色粉状固体1.2 g。收率:75.16%,mp:246℃~247℃;1H NMR (400 MHZ, DMSO-d6),δ = 2.42 (3H, S, CH3),7.35 (1H, d, J = 8.4 Hz, Ar-H),7.67 (1H, d, J = 7.2 Hz, Ar-H),7.77 (1H, t, J = 8.0 Hz, Ar-H),8.01 (1H, d, J = 1.6 Hz, Ar-H),8.54 (1H, d, J= 1.6 Hz, Ar-H),12.17 (1H, s, OH)。13C NMR (100 MHZDMSO-d6):δ = 20.67,116.40,118.91,124.45,125.11,126.82,130.14,132.33,134.99,137.04,137.07,150.17,161.37,164.75,168.82,180.44,186.58;IRν:3445,3074,1770,1691,1640,1609,1578,1459,1419,1372,1353,1331,1312,1278,1245,1225,1193,1155,1097,1075,1051,1021;ESI-MS m/z:Calcd for C17H10O7Na {[M-H]−}325.0427,found 325.042 6;Anal calcd for C17H10O7:C 62.34,H 3.32,O 34.46;found C 62.58,H 3.09,O 34.33。
2.2.5. 5-乙酰基-4-羟基-9,10-二氧-9,10-二氢蒽-2-羧酸(1b)的合成
将1.2 g化合物5加入到50 mL圆底瓶中,加入THF 12 mL,10%的Pd/C 0.12 g,加完氢气真空切换3次,后通氢气室温搅拌反应12小时,[TLC检测(展开剂:V (石油醚):V (乙酸乙酯):V(乙酸) = 20:10:1]。抽滤,滤液40℃减压蒸干得浅红棕色固体0.7 g,加入乙酸乙酯10 mL搅拌1小时后抽滤,滤饼5 mL乙酸乙酯淋洗一次,抽干,滤饼45℃减压干燥,得浅红棕色粉状固体0.5 g。收率:69.44%,mp:238℃~239℃;1H NMR (400 MHZ, DMSO-d6):δ = 2.41 (3H, S, CH3),7.60 (1H,d, J = 8.0 Hz, Ar-H),7.71 (1H, d, J = 7.2 Hz, Ar-H),7.96 (1H, d, J = 8.0 Hz, Ar-H),8.08 (1H, d, J = 1.2 Hz, Ar-H),8.13 (1H, d, J = 1.2 Hz, Ar-H),12.14 (1H, s, OH) 13CNMR (100 MHZ, DMSO-d6) δ = 20.67,118.39,118.67,124.02,124.13,125.21,130.56,132.85,134.53,136.14,137.94,150.01,161.06,165.15,168.80,180.60,186.77;IRν:3442,3096,2989,2853,2608,2545,1764,1705,1677,1645,1591,1567,1484,1445,1417,1368,1339,1270,1236,1205,1193,1155,1094,1045,1020,996。ESI-MS m/z:Calcd for C17H10O7Na{[M-H]−} 325.0427,found 325.0426;Anal calcd for C17H10O7:C 62.19,H 3.32,O 34.34;found C 62.58,H 3.09,O 34.33。
3. 结果与讨论
通过相关实验验证,本研究很容易得到双醋瑞因的两个关键杂质4-乙酰基-5-羟基-9,10-二氧-9,10-二氢蒽-2-羧酸和5-乙酰基-4-羟基-9,10-二氧-9,10-二氢蒽-2-羧酸;在混合物(1a, 1b)的制备过程中,主要产物为二乙酰大磺酸和(1a, 1b),其HPLC见图2。
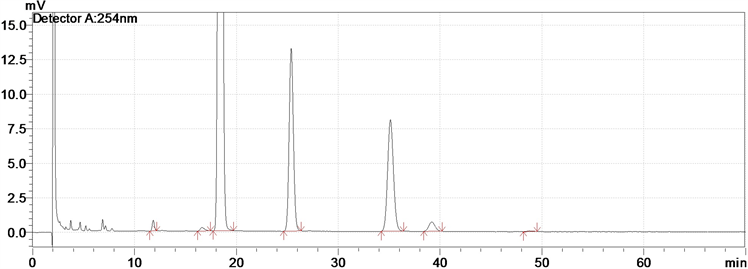
Figure 2. Mixture of HPLC
图2. 混合物HPLC图
[HPLC面积归一化法,色谱柱:用十八烷基键合硅胶(ZORBAX SB-C18)为填充剂(4.6 mm × 250 mm, 5 μm);流动相:甲醇-乙腈-0.005 mol/L磷酸二氢钾,用磷酸溶液调节至pH 1.6 (38:10:52);检测波长为254 nm;柱温为35℃;流速为1.0 mL/min。]
由于该混合物的化学结构决定其化学性质,并且1a、1b只是位置不一样,理化性质相似,溶解性也很差,在改变功能团来改变其理化性质的过程中使用过三甲基硅醚、叔丁基二苯基硅醚、叔丁醇和溴苄、对甲氧基溴苄、三苯甲基醚,通过实验验证前三个试剂保护时脱保护采用酸水解其酯建也会断裂,后三个保护试剂比较后溴苄效果最好。但是在用10%的Pd/C催化氢解制备1a、1b时,所使用的溶剂THF一定为无水,通过实验得出普通THF作为溶剂时产物为大黄酸3。
基金项目
贵州省化学药物开发利用工程实验室;贵州省普通高等学校药物化学工程研究中心(黔教合KY字[2014]219号)。
文章引用
代泽琴,张 毅,冯广卫. 双醋瑞因杂质的合成
Synthesis of Diacerein Impurities[J]. 化学工程与技术, 2020, 10(05): 362-366. https://doi.org/10.12677/HJCET.2020.105046
参考文献
- 1. 王霖, 毛昱嘉, 王文杰. 双醋瑞因对破骨细胞性骨破坏的抑制作用及机制[J]. 药学学报, 2006, 41(6): 555-560.
- 2. 吕茜茜, 夏冬辉, 李华. 荧光光谱法研究双醋瑞因与人血清白蛋白的相互作用[J]. 应用化学, 2011, 28(7): 836-840.
- 3. Tisserand, S., Baati, R., Nicolas, M., et al. (2004) Expedient Total Syntheses of RHE in and Diacerhein via Fries Rearrangement. Journal of organic Chemistry, 69, 8982-8983. https://doi.org/10.1021/jo049228l
- 4. 朱兴一, 郭巧凤, 吴雪玲. 合成双醋瑞因的新方法[J]. 化学合成, 2010,18(2): 269-270.
- 5. Di Napoli. (1994) A Process for the Preparation of Diacerein. EP0636602, 1994-07-07.
- 6. 马燕如, 赵肖玉, 徐正. 大黄酚和大黄酸的合成[J]. 化学合成, 2007, 15(2): 244-246.
- 7. 夏士朋. 大黄酸合成新工艺[J]. 四川化工与腐蚀控制, 2001, 5(4): 1-2.
- 8. Tisserand, S., Baati, R., Nicolas, M., et al. (2004) Expedient Total Syntheses of Rhein and Diaeerhein via Fries Rearrangement. Journal of Organic Chemistry, 69, 8982-8983. https://doi.org/10.1021/jo049228l
- 9. 黄巧巧, 陈雪帆, 李会林. 高效液相色谱法测定双醋瑞因含量与有关物质[J]. 医药导报, 2007, 26(9): 1077-1078.
- 10. Ashok, C., Golak, M., Adwait, D., et al. (2009) Isolation and Structural Elucidation of Two Impurities from a Diacere in Bulk Drug. Journal of Pharmaceutical and Biomedical Analysis, 49, 525-528. https://doi.org/10.1016/j.jpba.2008.11.015
- 11. 张毅, 王建塔, 汤磊. 双醋瑞因的合成及质量研究[J]. 化学试剂, 2014, 26(3): 273-275.
- 12. Gonnot, V., Tisserand, S., Nicolas, M., et al. (2007) Total Synthesis of Rhein and Di-acerhein via a Directed Ortho Metalation of an Aromatic Substrate. Tetrahedron Letters, 48, 7117-7119. https://doi.org/10.1016/j.tetlet.2007.07.218