Hans Journal of Medicinal Chemistry
Vol.04 No.04(2016), Article ID:19121,13
pages
10.12677/HJMCe.2016.44006
Progress in the Research of Multi-Target Tyrosine Kinase Inhibitors
Dan Liu*, Xue Li, Yi Zhang, Penghui Peng
College of Pharmaceutical and Biological Engineering, Shenyang University of Chemical Technology, Shenyang Liaoning

Received: Nov. 10th, 2016; accepted: Nov. 27th, 2016; published: Nov. 30th, 2016
Copyright © 2016 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



ABSTRACT
The study on multi-targeted tyrosine kinase inhibitors has become a new trend in researches on antitumor drugs in recent years. In order to offer reference to the research and development of tyrosine kinase inhibitors, this review introduced the research results of tyrosine kinase inhibitors with different structures and antitumor activity by summarizing related literature, at home and abroad.
Keywords:Multi-Target, Tyrosine Kinase Inhibitor, Antitumor

多靶点酪氨酸激酶抑制剂的研究进展
刘丹*,李雪,张毅,彭鹏辉
沈阳化工大学,制药与生物工程学院,辽宁 沈阳

收稿日期:2016年11月10日;录用日期:2016年11月27日;发布日期:2016年11月30日

摘 要
多靶点酪氨酸激酶抑制剂的研究已经成为近年来抗肿瘤药物研究的新趋势。本文整合国内外相关文献,综述了近年来各课题组对不同结构的酪氨酸激酶抑制剂及抗肿瘤活性的研究进展,以期为酪氨酸激酶抑制剂的研究与开发提供参考。
关键词 :多靶点,酪氨酸激酶抑制剂,抗肿瘤

1. 引言
目前癌症和心脑血管疾病已经占据全世界主要死因的前两位,虽然近年来对于癌症的相关研究已经取得了很大的进步,但是现有的抗肿瘤药物存在高毒性、易产生耐药性等缺点 [1] [2] [3] [4] 。因此研究新型靶向药物和满足全新治疗手段所需求的药物,已成为全球药业公司和各国政府的关注重点和未来研究发展的方向 [5] 。
人类基因组的数据表明,目前已知的蛋白激酶家族有518个成员 [6] [7] ,它们主要通过传递细胞内外的信号来调控细胞重要的生理过程。蛋白酪氨酸激酶(Protein tyrosine kinases (PTKs))作为蛋白激酶家族中非常重要的一员,PTKs将三磷酸腺苷(Adenosine Triphosphate, ATP)上的γ-磷酸基转移到底物的蛋白酪氨酸残基上,通过将酚羟基磷酸化来完成细胞间的信息传递,在细胞发育、调节和肿瘤细胞的分化、迁移、凋亡等过程中起着至关重要的作用 [8] [9] ,若PTKs在调节过程中失控将会影响其下游信号通路的正确激活,进而引起细胞增殖调节功能紊乱而引发许多疾病,如酪氨酸激酶活性过高使受体磷酸化进而激活下游信号,导致细胞过度转化、增殖、对抗细胞凋亡、促进细胞生存进而形成恶性肿瘤 [10] [11] [12] ,因此,将酪氨酸激酶作为新型靶点研制该类激酶抑制剂来抑制酪氨酸激酶的过度表达,恢复其生理平衡,已经成为一种新的抗肿瘤药物研究方向 [13] [14] [15] 。本文简要将蛋白酪氨酸激酶分为四大类,在此基础上分别介绍了近年来各课题组对多靶点酪氨酸激酶抑制剂的研究进展。希望能够以此为多靶点酪氨酸激酶抑制剂的相关研究与抗肿瘤药物开发提供参考。
2. 蛋白酪氨酸激酶的分类
人类基因组目前已发现约2000个激酶,其中蛋白酪氨酸激酶已超过90个。蛋白酪氨酸激酶家族主要以跨膜受体或胞质形式参与细胞信号转导过程,按照其结构、功能和在细胞内存在的位置大致可分为两类:受体酪氨酸激酶(receptor tyrosine kinase, RTK)和非受体酪氨酸激酶(nonreceptor tyrosine kinase, NRTK) [8] 。RTK均为单次跨膜运输,NRTK又可细分为:① 核内酪氨酸激酶,如Abl家族和Wee家族;② 胞质酪氨酸激酶,如Src家族、Tec家族、ZAP70家族、JAK家族 [16] 等。
2.1. 受体酪氨酸激酶
受体酪氨酸激酶(RTK)是一类可以使酪氨酸激酶磷酸化的穿膜受体蛋白 [17] ,通常具有以下结构:可以与配体相结合的胞外结构域、单次跨膜的疏水α螺旋区及含有RTK活性的细胞内结构域 [18] 。其配体一般为蛋白类激素以及膜结合或可溶性的多肽。含有RTK活性的细胞内结构区域即酪氨酸激酶的作用部位,该处具有自磷酸化位点,当其内在的催化活性被激活时,受体可以通过二聚化或寡糖化使这两个催化区域相结合,并转移活化口袋中的共生残基的磷酸,受体本身激酶活性将被激活进而自动磷酸化酪氨酸残基。若激酶过度表达,将有可能导致恶性肿瘤的形成 [19] 。
目前已知的58种受体酪氨酸激酶,根据细胞内外区域结构即受体/配体的不同可以被划分为20多个亚家族 [20] ,包括:表皮生长因子受体(epidermal growth factor receptor, EGFR)、血小板源生长因子受体(platelet-derived growth factor receptor, PDGF)、成纤维细胞生长因子受体(fibroblast growth factor receptor, FGFR)、血管内皮生长抑制因子受体(vascular endothelial growth factor receptor, VEGFR)、神经生长因子受体(nerve groeth factor receptor, NGFR)、原肌球蛋白受体激酶(Tropomyosin-related kinase, TRK)、肝细胞生长因子受体(hepatocyte growth factor receptor, HGFR)、红细胞生成素产生肝细胞受体(erythropoietin- producing hepatocellular receptor, EPHR)、血管生成素受体(TIE)、白细胞酪氨酸激酶(LTK)家族 [21] 等,它们在不同的细胞活动中发挥着非常重要的作用。
2.2. 非受体酪氨酸激酶
非受体酪氨酸激酶(NRTK)的调节机制差异较大,一般通过与细胞因子、生长因子或激素等跨膜受体发生物理作用进行胞外信号响应。经过长时间的研究发现,该类受体在某一条件下与胞外配体或黏着成分结合会使其活化。例如,在PDGFR激活Src的同时,与Src结构相似的酪氨酸激酶可与T细胞中T细胞抗原受体复合物、CD4或CD8抗原等相结合。HER-1在Src帮助下激活STAT因子;同理,JAK激酶(Janus kinase)家族在配体与细胞因子受体结合时被活化,进而磷酸化STAT因子,信号最终传导至细胞核内激活转录;IFN-γ也可通过与受体相结合发生磷酸化,进而与STAT相结合。在JAK的催化作用下,依照同样的活化机理,STAT二聚体转移到核内结合到DNA启动子的活化序列上,诱导靶基因表达从而促进合成多种蛋白质,提高了细胞抗病毒活性 [22] 。
3. 多靶点小分子酪氨酸激酶抑制剂发展概况
酪氨酸激酶抑制剂(TKIs)是小分子抑制剂,能够渗透过细胞膜并靶向肿瘤细胞和周围内皮和血管激酶受体的特定部位,从而阻断细胞增殖信号传导途径 [23] 。TKIs从结构上大致可以分为以下几大类:喹唑啉类、喹啉类及苯胺、苯酰胺类等,这些抑制剂的母核大多模拟ATP中腺嘌呤结构即含有含氮原子杂环的同时结构中都有一个或多个杂原子。它们分别对特定或多个酪氨酸激酶具有专一或选择性抑制作用 [24] 。
3.1. 喹唑啉类酪氨酸激酶抑制剂
目前,经 FDA审核批准了一批 EGFR 小分子靶向抑制剂,大多数属于喹唑啉类——已发现的选择性最好、活性最高的要属苯胺喹唑啉类化合物,其主要作用于EGFR,目前已有数个该类抑制剂上市,还有大量正在开发或已经进入临床的化合物 [25] [26] 。为了提高活性一般采用以下形式对结构进行改造:在苯胺3位取代基添加亲脂性取代基,将苯胺上的H替换为F、Cl、Br或I原子;在喹唑啉的6、7位上连接给电子基团;在喹唑啉环上的7位取代位置引入N、O原子 [27] 。目前上市的各种含喹唑啉基团的酪氨酸激酶抑制剂的结构均符合以上构效关系。
AstraZeneca [28] 公司开发的苯胺喹唑啉类化合物——吉非替尼(Gefitinib, ZD-139)可以竞争结合EGFR-TK催化区上Mg2+-ATP,选择性抑制ErbB受体同时也可以抑制有丝分裂原活化蛋白激酶的活化。在异种移植模型中,对降低肿瘤细胞生长速度、缩小甚至使其生长停滞起很大作用,经IDEAL-1 (Iressa Dose Evaluation in Advanced Lung Cancer)研究 [29] 对其250 mg∙d−1和500 mg∙d−1剂量组治疗进行安全性和缓解性测试结果显示有效率分别为18.4%和19.0%,IDEAL-2 [30] 实验结果显示有效率分别为11.8%和8.8%,不良反应率为6.9%和17.5%,两组的症状缓解率分别为43%和34%,该药物通过口服可以广泛分布于身体组织和器官中,药物生物利用率高达50%~60%,2002年首次于日本上市,2003年5月作为三线单一治疗药物在美国及澳大利亚获准用于治疗晚期非小细胞肺癌,常见副反应为皮疹、粉刺、腹泻、恶心、呕吐等 [31] 。
Enetech公司与OSI公司联合开发的厄洛替尼(Erlotinib, Genentech)是一种以EGFR为靶点的小分子酪氨酸酶抑制剂。体内体外实验资料表明Erlotinib可以特异地抑制EGFR激酶活性(IC50 = 2 μM),对比其他酪氨酸激酶抑制剂相比效果显著。经Ⅰ、Ⅱ、Ⅲ期临床试验结果显示,它可以明显延长患者平均生存期效果优于同期另一药物吉非替尼,主要副反应为头痛、疲劳、恶心、呕吐、腹泻及痤疮样皮疹。2002年进入FDA新药审批程序,两年后获得美国FDA批准用于治疗一线化疗失败的局部晚期或转移性非小细胞肺癌 [32] ,目前已经在瑞士、德国、英国、爱尔兰等国上市。
李冬冬 [33] 等设计并合成了一系列靶向EGFR酪氨酸激酶的喹唑啉骨架肉桂酰胺类衍生物(1-25) (见表1),并初步测试了它们对肿瘤细胞抗增殖活性和体外EGFR激酶抑制活性。其中大部分化合物表现出对非小细胞肺癌细胞A549和小鼠黑色素瘤细胞B16-F10细胞抗增殖活性。经构效分析和优化,得到活性较好的两种化合物24和25,它们对EGFR激酶的抑制活性IC50值分别为0.12 μM和0.19 μM。
HU [34] 等人报道了25个作为C-Kit的强效抑制剂的化合物,它们都属于芳氨基喹唑啉类,抑制作用是蛋白酪氨酸磷酸酶(Src)、丝裂原活化蛋白激酶(p38)、淋巴细胞特异性蛋白酪氨酸激酶(Lck)和血管内表皮生长因子受体(KDR)的200倍,具有理想的药代动力学特性.通过在啮齿类动物肥大细胞活化的药效学模型实验证明了化合物26 (见图1)具有成为抗肝癌药物的潜质。
VANBROCKLIN [35] 等通过EGFR酪氨酸激酶放射性实验证明了苯胺二烷氧基喹唑啉化合物对EGFR酪氨酸激酶有一定的亲和性,可作为筛选表皮生长因子受体酪氨酸激酶抑制剂的潜在的肿瘤成像探针。化合物4-(2’-氟苯氨基)-6,7-二乙氧基喹唑啉(27)、4-(3’-氟苯氨基)-6,7-二乙氧基喹唑啉(28)、4-(2’-氯苯氨基)-6,7-二甲氧基喹唑啉(29)、4-(3’-溴苯氨基)-6,7-二甲氧基喹唑啉30) (见图2)与EGFR酪氨酸激酶有较强的结合能力。
Fatmah [36] 等报道了一系列噻唑并[2,3-b]喹唑啉类化合物(化合物31-34) (见图3),体外抗癌活性测试表明这四个化合物具有广谱的抗癌活性。化合物31,32的平均IC50、TGI、LC50值分别为2.5、>100、>100 μM和2.4、9.1、36.2 μM,活性是氟尿嘧啶的9倍。
Chen [37] 等以埃罗替尼为先导化合物,设计并合成了8种新型三嗪并喹唑啉类化合物(化合物35-42),如图4,和埃罗替尼相比,这8种化合物具有更高的抗癌活性,其中化合物39和40对N87、A431、H1975、BT474和Calu-3 5种癌细胞系的EGFR的IC50都为0.043 μM经过更进一步研究可以发现含有嗪环的化合物比噁嗪环化合物具有更高的抗癌活性。
3.2. 喹啉类酪氨酸激酶抑制剂
含喹啉结构的酪氨酸激酶抑制剂是在喹唑啉类化合物的构效关系基础上设计开发的。研究发现,对比喹唑啉类化合物,为了获得更好的构型和更稳定的电荷分布可以将喹唑啉3位上的N替换为C并引入吸电基团而对其活性并不产生影响。
伯舒替尼(Bosutinib, SKI-606)是由Wyeth公司开发的3-氰基喹啉类双重抑制剂,作用靶点为SRC和ABL。主要用于治疗非小细胞肺癌、乳腺癌等实体瘤和急性淋巴性白血病、慢性粒细胞白血病等。对SRC的IC50值为1.2 μM,它的抑制性作用很强,对成纤维细胞的IC50值为100 μM [38] 。
DAN LIU [39] 等在吉非替尼的基础上设计并合成了22个新型喹啉类酪氨酸激酶抑制剂(化合物43-64),以Gefitinib作为参照,其中化合物43、48和59对HeLa和BGC-823癌细胞系具有显著的抑制活性(见表2),其IC50值分别为17.92、10.18、7.15 μM和3.63、8.32、4.65 μM。
LIU [40] 通过在结构上进行新型药物设计的方法,设计合成了以BMX激酶为靶点的小分子化合物65 (见图5),它可以很好地抑制重组BMX激酶(IC50 = 8 μM),作为目前为止首个被报道的高选择性BMX激酶抑制剂,它的出现为以后针对BMX激酶的抗癌药物的研发奠定了基础。

Table 1. The chemical structures of compounds 1-25
表1. 化合物1-25的化学结构
 table_table_table_3-3070030_1_hanspub.htm
table_table_table_3-3070030_1_hanspub.htm
Figure 1. The chemical structures of compounds 26
图1. 化合物26的结构
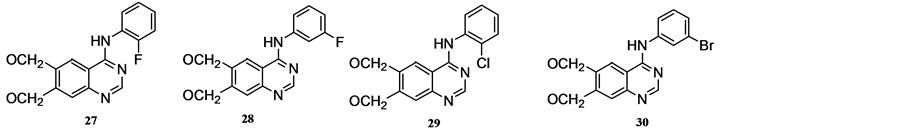
Figure 2. The structure of compound 27-30
图2. 化合物27-30的结构
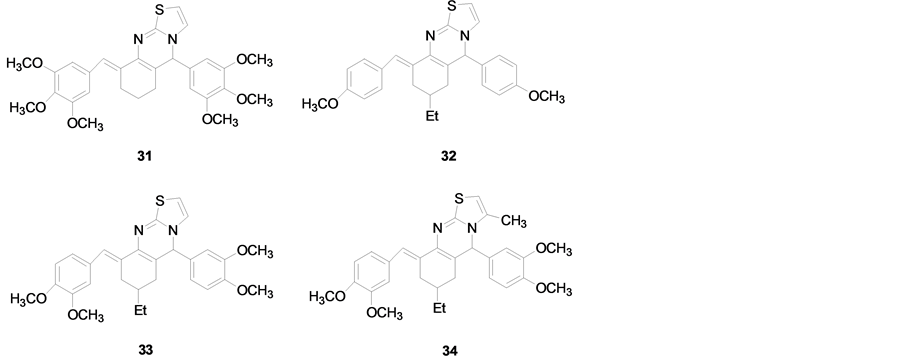
Figure 3. The structure of compound 31-34
图3. 化合物31-34的结构
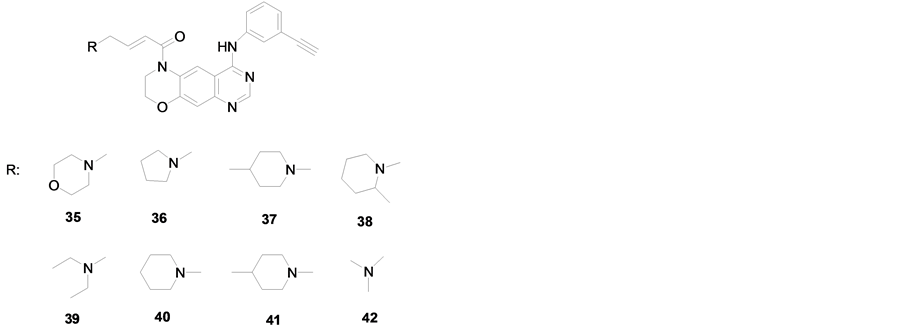
Figure 4. The structure of compound 35-42
图4. 化合物35-42的结构

Figure 5. The structure of compound 65
图5. 化合物65的结构

Table 2. Antiproliferative activity of the target compounds on Hela and BGC-823 cell lines
表2. 目标化合物对Hela和BGC-823细胞系的抑制活性
aInhibitory percentage of cells treated with each compound at a concentration of 10 μM for 96 h; bThe agent concentration that inhibited Hela and BGC-823 cells growth by 50%.
李伟 [41] 等通过研究一系列酪氨酸激酶抑制剂的构效关系,利用计算机辅助药物设计手段以及生物电子等排、骨架迁越原理等设计合成了一系列多靶点喹啉类衍生物(见表3化合物66-85),其中化合物66抑制活性最强,对HeLa,Eca-109及A549细胞的IC50值分别为(1.04 ± 0.12) (0.37 ± 0.06) (2.16 ± 1.05) μM,对肿瘤细胞的抑制活性强于舒尼替尼。

Table 3. The chemical structures of compounds 66-85
表3. 化合物66-85的化学结构
Morphy [42] 等在伯舒替尼的结构基础上进行结构修饰,在其4-苯氨基位置引入2-氨基噻唑基团,获得的化合物86 (见图6)对Src有很好的抑制作用,其IC50值为15 μM。
3.3. 苯胺、苯酰胺类酪氨酸激酶抑制剂
张晶 [43] 等以4-氯-6,7-二甲氧基喹啉、6-氯-7氮杂吲哚、4-氯-7-氮杂吲哚为原料与具有多种生物活性的优势结构噻吩环、呋喃环、及邻甲氧基苯环进行连接,经取代、还原、缩合反应合成9个酰胺类化合物(见图7化合物87-95)。并对其进行体外抗肿瘤活性筛选,结果显示化合物87、88、89在GTL-16细胞上有较好的药效活性。
刘长春 [44] 等在对达沙替尼的研究基础上设计合成了2-氨基-N-(1-氯-6-甲基苯基)噻唑-5-甲酰胺(见图8化合物96),该化合物对诱生型NO合成酶有选择抑制作用 [38] 。

Figure 6. The structure of compound 86
图6. 化合物86的结构

Figure 7. The structure of compound 87-95
图7. 化合物87-95的结构

Figure 8. The structure of compound 96
图8. 化合物96的结构
3.4. 其他类型
GANGNJEE [45] 等设计合成的6种新颖N4-取代苯基-6-取代苯甲基-7H-吡咯并[2,3-d]嘧啶-2,4-二胺可作为多种受体酪氨酸激酶抑制剂(见图9),其中化合物97、98、99对多种RTK显示出强有效的抑制作用,化合物100和101对VEGFR-2 ((0.048 ± 0.06)和(0.1 ± 0.021) μM)的强效抑制作用明显超过了PDGFR-β ((193.2 ± 20.1)和(145 ± 23.8) μM)及VEGFR-1 (>200和(185.6 ± 27.5) μM)。
LANG [46] 等设计合成的5个9-苯乙氨基吖啶类化合物102-106可以作为潜在的酪氨酸激酶和拓扑异构酶Ⅰ的多靶点抑制剂,对人肝癌细胞HEPC-2显示强抗增殖活性,其中化合物106对VEGFR-2和Src具有抑制活性(见表4、表5)。
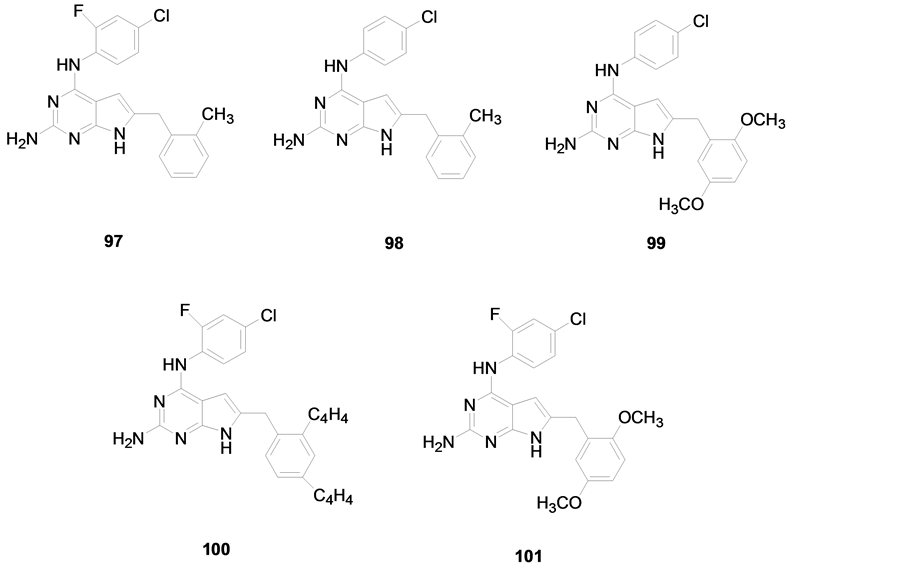
Figure 9. The structure of compound 97-101
图9. 化合物97-101的结构

Table 4. The structure and antiproliferative activity of the target compounds on HepG-2 cell lines
表4. 目标化合物结构及对HepG-2细胞系的抑制活性
Table 5. Percent inhibition effect of compounds 105 and 106 selected at 20 μM on the activity of two kinases
表5. 化合物105和106在20 μM时对Src和VEGFR-2两种酶的抑制率
4. 展望
酪氨酸激酶在肿瘤细胞的生长和增殖中起着非常重要的作用,在过去的几年里出现了数个以蛋白酪氨酸激酶抑制剂为靶向的抗肿瘤药物,这些药物克服了传统药物选择性差、副作用大等缺点,具有高选择性、低毒性的治疗效果。由于肿瘤增殖分化作用机制十分复杂,因此单一靶点药物难以在肿瘤治疗方面取得较好的效果,因此设计合成低毒、高选择性、高效的多靶点的酪氨酸激酶抑制剂也是抗肿瘤新药研发的主要方向。虽然酪氨酸激酶小分子抑制剂的开发取得了上述的很多进展, 但也面临着很多挑战:① 肿瘤细胞基因突变所造成的激酶抑制剂耐药问题日益严重;② 如何在多靶点酪氨酸激酶抑制剂和提高激酶选择性的问题上寻找到一个平衡点;③ 如何在进入临床前对化合物毒性、药动学性质等做出比较准确的预测,以提高新药开发的速率和避免重复浪费。因此,全面有效地开发抗肿瘤药物需要各领域的学者通力协作,可以预见,设计合成酪氨酸激酶小分子抑制剂治疗肿瘤疾病必将走入一个新的时代。相信在不久的将来,小分子酪氨酸激酶抑制剂将克服肿瘤多药耐药性,实现多靶点抑制的开发,实现小分子TKI安全性评价和个体化给药,在癌症治疗领域发挥更重要的作用。
文章引用
刘 丹,李 雪,张 毅,彭鹏辉. 多靶点酪氨酸激酶抑制剂的研究进展
Progress in the Research of Multi-Target Tyrosine Kinase Inhibitors[J]. 药物化学, 2016, 04(04): 42-54. http://dx.doi.org/10.12677/HJMCe.2016.44006
参考文献 (References)
- 1. Fukuoka, M., Yano, S., Giaccone, G., et al. (2003) Multi-Institutiongal Randomized Randomized PhaseⅡ Trial of Gefitinib for Previously Treated Patients with Advanced Non-Small-Cell Lung Cancer. Journal of Clinical Oncology, 21, 2237-2246.
- 2. Kris, M.G., Natale, R.B., Herbst, R.S., et al. (2003) Efficacy of Gefitinib, an Inhibitor of the Epidermal Growth Factor Receptor Tyrosine Kinase, in Symptomatic Patients with Non-Small Cell Lung Cancer—A Randomized Trial. JAMA, 290, 2149-2158. https://doi.org/10.1001/jama.290.16.2149
- 3. 王慧杰, 王燕, 张湘如, 等. Gefitinib靶向治疗非小细胞肺癌的现状与进展[J]. 癌症进展杂志, 2005, 3(2): 163- 169.
- 4. Dong, Q.G. (2004) Epidermal Growth Factor Receptor Family and Molecular Targeting Therapy of Lung Cancer. Cancer, 24, 93-95.
- 5. 李冬冬. 喹唑啉类化合物的设计, 合成及生物活性评价[D]: [博士学位论文]. 南京: 南京大学, 2014.
- 6. Hu, E., Tasker, A., White, R.D., et al. (2008) Discovery of Aryl Aminoquinazoline Pyridones as Potent, Selective, and Orally Efficacious Inhibitors of Receptor Tyrosine Kinase C-Kit. Journal of Medicinal Chemistry, 51, 3065-3069. https://doi.org/10.1021/jm800188g
- 7. Vanbrocklin, H.F., Lim, J.K., Coffing, S.L., et al. (2005) Anilinodialkoxyquinazolines: Screening Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors for Potential Tumor Imaging Probes. Journal of Medicinal Chemistry, 48, 7445-7456. https://doi.org/10.1021/jm050607w
- 8. 郭建军, 朱晶, 赵永跃, 等. 不可逆性酪氨酸激酶抑制剂的研究进展[J]. 中国药理学通报, 2015, 31(6): 749-754.
- 9. Al-Omary, F.A.M., Hassan, G.S., El-Messery, S.M., et al. (2012) Subsitituted Thiazoles V. Synthesis and Antitumor Activity of Novel Thiazolo [2,3-b] Quinazoline and Pyrido [4,3-d]Thiazolo[3,2-d] Pyrimidine Analogues. European Journal of Medicinal Chemistry, 47, 65-72 https://doi.org/10.1016/j.ejmech.2011.10.023
- 10. HU, S.J., XIE, G.J., ZHANG, D.X., et al. (2012) Synthesis and Biological Evaluation of Crown Ether Fused Quinazoline Analogues as Potent EGFR Inhibitors. Bioorganic & Medicinal Chemistry Letters, 22, 6301-6305. https://doi.org/10.1016/j.bmcl.2012.06.067
- 11. Jallal, H., Valentino, M.L., Chen, G., et al. (2007) A Src/Abl Kinase Inhibitor, SKI-606, Blocks Breast Cancer Invasion Growth and Metastasis in Vitro and in Vivo. Cancer Research, 67, 1580-1588. https://doi.org/10.1158/0008-5472.CAN-06-2027
- 12. Dan, L., Tian, L., Jian, K., Ying, Z. and Hai, F.W. (2016) Synthesis and Anti-Tumor Activities of 4-Anilinoquinoline Derivatives. Molecules, 21, 21.
- 13. Liu, F.Y. (2013) Discovery of a Selective Irreversible BMX Inhibitor for Prostate Cancer. ACS Chemical Biology, 8, 1423-1428. https://doi.org/10.1021/cb4000629
- 14. Li, W., Lei, C.H., Wang, L.Q., et al. (2016) Synthesisi and Preliminary Research of Quinoline Derivatives as Multi- Targeted Receptor Tyrosine Inhibitors. Chinese Journal of New Drugs, 25, 1522-1529.
- 15. Morphy, R. (2010) Selectively Nonselective Kinase Inhibition: Striking the Right Balance. Journal of Medicinal Chemistry, 53, 1413-1437. https://doi.org/10.1021/jm901132v
- 16. 张晶, 程璐, 梁泰刚, 等. 苯酰胺类C-Met激酶抑制剂的合成及其体外抗肿瘤活性研究[J]. 现代药物与临床, 2016, 31(9): 1313-1318.
- 17. 刘长春, 王天扬, 张雨晴, 等. 2-氨基-N-(2-氯-6-甲基苯基)噻唑-5-甲酰胺的合成[J]. 中国医药工业杂志, 2016, 47(7): 848-850.
- 18. Gangjee, A., Kurup, S., Ihant, M.A., et al. (2012) N4-Aryl-6-Substitutedphenylmetyl-7-H-Pyrrol[2,3-d] Pyrimidine-2, 4-Diamine as Receptor Tyrosine Kinase Inhibitors. Bioorganic & Medicinal Chemistry, 20, 910-914. https://doi.org/10.1016/j.bmc.2011.11.058
- 19. 康从民, 赵绪浩, 于玉琪, 等. 吲哚衍生物类VEGFR-2酪氨酸激酶抑制剂的从头设计[J]. 高等学校化学学报, 2014, 35(3): 550-554.
- 20. 宋艳宁, 张赫然, 尹东东, 等. 小分子酪氨酸激酶抑制剂在癌症靶向治疗的研究进展[J]. 中国药学杂志, 2016, 51(3): 165-169.
- 21. 王乐, 李明春, 等. 表皮生长因子受体酪氨酸激酶抑制剂联合用药治疗非小细胞癌的研究进展[J]. 中国药理学与杂志, 2016, 51(3): 165-169.
- 22. Giamas, G., Man, Y.L., Hirner, H., et al. (2010) Kinases as Targets in the Treatment of Solid Tumors. Cell Signal, 22, 984-1002. https://doi.org/10.1016/j.cellsig.2010.01.011
- 23. 李文丽. 酪氨酸蛋白激酶抑制剂的设计、合成和筛选[D]: [硕士学位论文]. 吉林: 吉林大学药学系, 2014.
- 24. 王勇, 龙亚秋. 蛋白酪氨酸激酶小分子抑制剂的研究新进展[J]. 有机化学, 2011, 31(10): 1595-1606.
- 25. 茆勇军, 李海泓, 李剑峰, 等. 蛋白酪氨酸激酶信号转导途径与抗肿瘤药物[J]. 药学学报, 2008, 43(4): 323-334.
- 26. 刘靖, 王林, 杨晓明, 等. 多靶点蛋白酪氨酸激酶抑制剂的研究进展[J]. 国际药学研究杂志, 2009, 36(3): 161- 163.
- 27. Summy, J.M. and Gallick, G.E. (2006) Treatment for Advanced Tumors: SRC Reclaims Center Stage. Clinical Cancer Research, 12, 1398-1401.
- 28. Schenone, S., Manetti, F. and Botta, M. (2007) SRC Inhibitors and Angiogenesis. Current Pharmaceutical Design, 13, 2118-2128.
- 29. Weisberg, E., Manley, P.W. and Cowan-Jacob, S.W. (2007) Second Generation Inhibitors of BCR-ABL for the Treatment of Imatinib-Resistant Chronic Myeloid Leukaemia. Nature Reviews Cancer, 7, 345-356. https://doi.org/10.1038/nrc2126
- 30. Schenone, S., Manetti, F. and Botta, M. (2007) Last Findings on Dual Inhibitors of abl and SRC Tyrosine-Kinases. Mini Reviews in Medicinal Chemistry, 7, 191-201.
- 31. Cao, X., You, Q.D. and Li, Z.Y. (2008) Recent Progress of SRC Family Kinase Inhibitors as Anticancer Agents. Mini Reviews in Medicinal Chemistry, 8, 1053-1063.
- 32. Diane, H.B. (2008) Exploitation of the 3-Quinolinecarbonitrile Template for SRC Tyrosine Kinase Inhibitors. Current Topics in Medicinal Chemistry, 8, 922-934.
- 33. 张秋荣, 陈婷, 于康, 等. 多靶点蛋白酪氨酸激酶抑制剂类抗肿瘤药物的研究进展[J]. 海峡药学, 2012, 24(2): 8.
- 34. 康从民, 代英杰, 王巧燕, 等. 小分子蛋白酪氨酸激酶抑制剂及其作用机制[J]. 中国新药杂志, 2013, 22(10): 1170-1172.
- 35. 王洪波, 陈晓光, 等. EGFR抑制剂耐药机制研究的新进展[J]. 国际药学研究杂志, 2007, 34(5): 347-355.
- 36. 王灵杰, 胡雅芳, 汪凤梅, 等. 多靶点酪氨酸激酶抑制剂的研究进展[J]. 中国药房,2013, 24(13): 1233-1235.
- 37. 宋艳宁, 张赫然, 尹东东, 等. 小分子酪氨酸激酶抑制剂在癌症靶向治疗的研究进展[J]. 中国药学杂志, 2016, 51(3): 165-171.
- 38. 张秋荣, 陈婷, 于康, 等. 多靶点蛋白酪氨酸激酶抑制剂类抗肿瘤药物的研究进展[J]. 海峡药学, 2012, 24(2): 7-10.
- 39. 吴建楠, 宋萌. 非受体酪氨酸激酶对细胞功能的影响及其研究进展[J]. 实用医学杂志, 2015, 31(3): 339-340.
- 40. Zhang, J., Yang, P.L. and Gray, N.S. (2009) Targeting Cancer with Small Molecule Kinase Inhibitors. Nature Reviews Cancer, 9, 28-39. https://doi.org/10.1038/nrc2559
- 41. 李伟, 雷春花, 王立强, 等. 多靶点受体酪氨酸激酶抑制剂喹啉衍生物的合成及活性初步研究[J]. 中国新药杂志, 2016, 25(13): 1522-1530.
- 42. Liu, D., Luan, T., et al. (2014) Advance in Search for Afatinib and Its Aalogues, the Multi-Target Tyrosine Kinae Inhibitor. Chinese Pharmaceutical Journal, 49, 2145-2149.
- 43. Khan, I., Ibrar, A., Ahmed, W. and Saeed, A. (2015) Synthetic Approaches, Functionalization and Therapeutic Potential of Quinazoline and Quinazolinone Skeletons: The Advances Continue. European Journal of Medicinal Chemistry, 90, 124-169. https://doi.org/10.1016/j.ejmech.2014.10.084
- 44. 黄庄霖. 喹唑啉结构的多靶点酪氨酸激酶抑制剂研究进展[J]. 海峡药学, 2011, 23(10): 22-24.
- 45. Han, W., Pan, H., Chen, Y., et al. (2011) EGFR Tyrosine Kinase Inhibitors Activate Autophagy as a Ytoprotrctive Response in Human Lung Cancer Cells. PLoS ONE, 6, e18691. https://doi.org/10.1371/journal.pone.0018691
- 46. Lang, X., Sun, Q., Chen, Y., et al. (2013) Novel Synthetic 9-Benzyloxyacridine Analogue as Both Tyrosine Kinase and Topoisomerase l Inhibitor. Chinese Chemical Letters, 44, 661-665. https://doi.org/10.1016/j.cclet.2013.05.018
*通讯作者。