Asian Case Reports in Pediatrics
Vol.1 No.1(2013), Article ID:9570,4 pages DOI:10.12677/ACRP.2013.11001
Clinical and MRI Analysis of Merosin-Deficient Congenital Muscular Dystrophy Type 1A
Department of Children’s Neuro-Endocrinology, Sun Yat-sen Memorial Hospital of Sun Yat-sen University, Guangzhou
Email: hobbyvan@163.com
Received: Dec. 11th, 2012; revised: Dec. 15th, 2012; accepted: Jan. 14th, 2013
ABSTRACT:
Objective: To explore the clinical and MRI features of Merosin-deficient congenital muscular dystrophy type 1A (MDC1A). Methods: The data of clinic and MRI of MDC1A (n = 8) reported in china were analyzed retrospectively. Results: Of the 8 cases, there were four males and four females. All cases presented with motor development retardation, muscular weakness and severe hypotonia within the first 6 months since birth, but without eye abnormalities. The levels of serum creatine kinase (CK) were elevated markedly. Electromyography suggested a myogenic damage process. Immunohistochemical studies of muscle biopsies for merosin were negative, and the ones for alpha, beta-dystroglycan were positive. The main MRI findings of MDC1A (n = 8) were observed as follows: symmetrical white matter abnormalities with hyperintensity on MRI T2 weighted and hypointensity on MRI T1 weighted. Conclusion: The diagnosis of MDC1A is based on the clinical findings of severe congenital hypotonia, weakness with high blood levels of creatine kinase, white matter abnormalities, and dystrophy associated with negative immunostaining of biopsied muscle for merosin.
Keywords: Merosin-Deficient Congenital Muscular Dystrophy Type 1A; Laminin Alpha-2; MRI; White Matter
Merosin缺陷性先天性肌营养不良的临床特征及其MRI特点(附1例报道)
何展文,罗向阳,梁立阳,李平甘,岑丹阳,李栋方,孟 哲
中山大学孙逸仙纪念医院儿童神经内分泌专科,广州
Email: hobbyvan@163.com
摘 要:
目的:探讨Merosin缺陷性先天性肌营养不良的临床特征和头颅MRI特点。方法:通过回顾性分析我国报道Merosin缺陷性先天性肌营养不良临床资料,总结Merosin缺陷性先天性肌营养不良的临床特征和MRI特点。结果:8例Merosin缺陷性先天性肌营养不良,男4例,女4例。发病年龄都在出生后6个月内,主要临床表现为运动发育迟缓,肌无力,无眼部病变。肌酸肌酶(CK)显著升高。肌电图检查示肌源性损害。肌肉免疫组化示Merosin染色阴性,α、β抗肌萎缩相关糖蛋白染色阳性。8例患儿头颅MRI检查示双侧大脑半球白质对称性异常信号,呈T1低信号,T2呈高信号。结论:Merosin缺陷性先天性肌营养不良主要根据先天性肌张力低下、肌无力临床表现,CK显著升高,脑白质异常信号和肌肉活检免疫组化Merosin染色阴性明确诊断。
收稿日期:2012年12月11日;修回日期:2012年12月15日;录用日期:2013年1月14日
关键词:Merosin缺陷性先天性肌营养不良;层粘连蛋白α2;MRI;脑白质
1. 引言
Merosin缺陷性先天性肌营养不良(Merosin-deficient congenital muscular dystrophy type 1A, MDC1A)是层粘连蛋白α2链缺陷的先天性肌营养不良[1]。本病欧美国家较为多见,国内对该病报道较少,至今国内报道的Merosin缺陷性先天性肌营养不良病例仅有7例[2-4]。为了提高临床医生对本病的认识,现将我院收治的1例Merosin缺陷性先天性肌营养不良报告如下,并复习文献结合我国报道病例的临床和MRI资料作回顾性分析,总结Merosin缺陷性先天性肌营养不良的临床特征和MRI特点。
2. 本科1例病例报道
病史。患儿,女,3岁,因“发现运动发育迟缓,肌酶增高2年半”于2010.10.18入院。出生后6个月家人发现患儿运动发育迟缓,颈软,不会抬头,不会学坐,在外院查肌酶提示肌酸肌酶(CK)3008U/L,肌酸肌酶同工酶(CK-MB)235 U/L,甲状腺功能正常,尿遗传代谢病筛查示:轻度乳酸尿伴半乳糖增高。脑电图无异常。SMN1基因检查无异常。曾在外院予康复锻炼、针灸治疗。患儿1岁2个月能抬头,至今仍坐立不稳,不能站立,1岁会说话,语言发育正常。患儿足月剖宫产出生,出生体重3.1 kg,否认有窒息抢救病史。母孕期无特殊。否认家族有类似疾病史。
体格检查。入院体查:发育尚可,智力、语言发育正常,视力检查及眼球运动正常,心肺腹检查无特殊。脑神经检查未见异常。颈无抵抗,克氏征(-),巴氏征(-)。双侧腓肠肌无肥大,骨关节无挛缩。四肢肌张力减低,双上肢肌力Ⅳ级,双下肢肌力Ⅲ级,腱反射减弱,深浅感觉正常。
辅助检查。血常规:WBC8.91 × 109/L,N0.55,RBC4.41 × 1012/L,Hb115 g/L,Plt294 × 109/L.;尿、粪常规正常;生化:谷丙转氨酶(ALT)39U/L,谷草转氨酶(AST)10 U/L,CK1556 U/L,乳酸脱氢酶(LDH)445 U/L,CK-MB 98 U/L,空腹血糖4.8 mmol/L,总胆固醇4.78 mmol/L,甘油三脂0.88 mmol/L,高密度胆固醇1.19 mmol/L,低密度胆固醇3.58 mmol/L,尿酸223 μmol/L,血氨10.0 μmol/L。致畸四项阴性。血尿遗传代谢筛查未见异常。血淋巴细胞亚群:B淋巴细胞亚群CD19 + 28.4%,CD20 + 27.3%,均明显升高;而T淋巴细胞亚群和血清免疫球蛋白未见异常。头颅MRI示双侧额叶、顶叶、颞叶及枕叶白质内见大片样异常信号区,边界不清,在T1W1上为稍低信号,在T2W1上为稍高信号,增强扫描强化不明显。双侧基底节区未见明显异常。小脑未见异常。脑室系统未见明确异常。各脑回未见异常。MRS示:双侧脑白质内ROV的NAA峰相对Cho峰和Cr峰略低,Lac峰未见明确增高(如图1)。分子遗传学检查LAMA2基因65个外显子,发现c.7147 C > T(杂合)为无义突变(p. Arg 2383X),此突变影响LAMA2基因功能,最终确诊为Merosin缺陷性先天性肌营养不良。
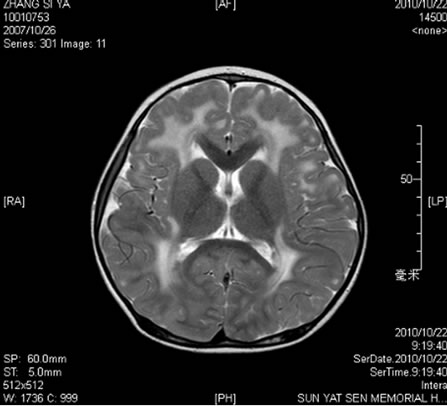
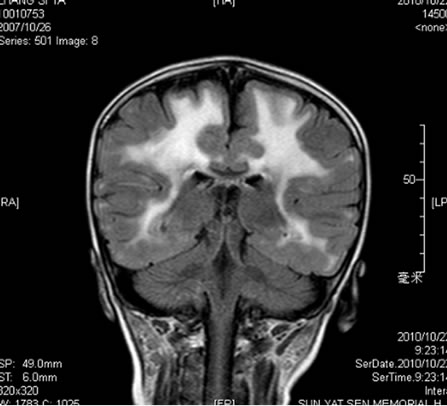
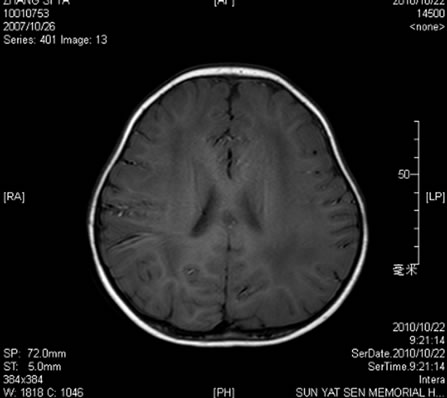
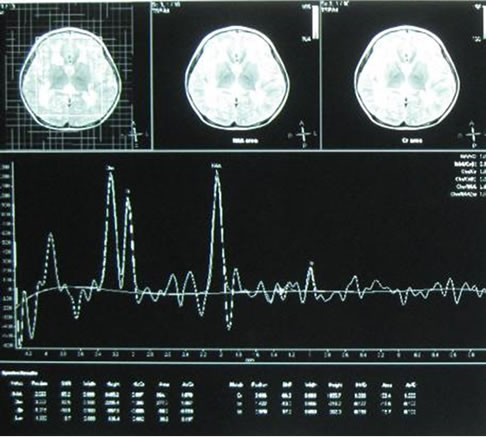
Figure 1. MRI of Merosin-deficient congenital muscular dystrophy type 1A at the age of 3 years
图1. Merosin缺陷性先天性肌营养不良MRI表现
治疗及随访。患儿治疗上予营养肌肉、改善循环、康复理疗基础上配合免疫调节治疗8个月,患儿运动功能逐渐进步,患儿现能坐稳,扶站,无运动倒退。
3. 国内报道8例Merosin缺陷性先天性肌营养不良的临床资料(表1)
一般情况。8例患儿中,男4例,女4例,男女比例为1:1,所有患儿均在出生时或出生后6个月内发病。
临床症状和体征。8例患儿主要临床表现为运动发育落后,抬头、独坐延迟,大部分患儿都不能独站和走路;2例患儿智力低下,2例合并癫痫,1例合并有胸廓畸形。患儿均都无眼部和听力症状。家族史均无特殊。体格检查8例患儿都有不同程度肌力下降,肌张力降低,腱反射减弱或消失。4例患儿合并关节挛缩,1例有肌肉假性肥大。
实验室检查。8例患儿进行血清CK检查,CK都中度或显著升高,其他LDH、CK-MB都有不同程度升高。7例患儿行肌电图检查,肌电图检查示肌源性损害。7例患儿行肌肉活检,肌肉病理有肌纤维的坏死、再生,肌间纤维结缔组织增加,均符合“肌营养不良”改变:肌纤维大小不等,见较多小圆状纤维发育不良的肌纤维及代偿肥大的纤维,肌周核增生伴轻度核内移,肌束内纤维结缔组织显著增生,肌纤维无明显坏死及新生。6例患儿行肌肉免疫组织化学染色,6例merosin染色阴性,6例α抗肌萎缩相关糖蛋白染色阳性,6例β抗肌萎缩相关糖蛋白染色阳性。
MRI结果。8例患儿均行头颅MRI检查,所有病例都表现有双侧大脑半球白质对称性病变,呈T1低信号,T2呈高信号;2例患儿MRI有脑室系统扩大,2例患儿MRI有小脑内囊肿,1例患儿MRI有脑萎缩;所有患儿内囊、胼胝体、基底节和丘脑均无受累。1例患儿行头颅MRS检查,双侧脑白质内ROV的NAA峰相对Cho峰和Cr峰略低,Lac峰未见明确增高。
4. 讨论
先天性肌营养不良(congenital muscular dystrophy, CMD)是一组异质性遗传性神经肌病,患儿多在出生时或生后数月内起病,以肌张力低下为表现,伴(或)
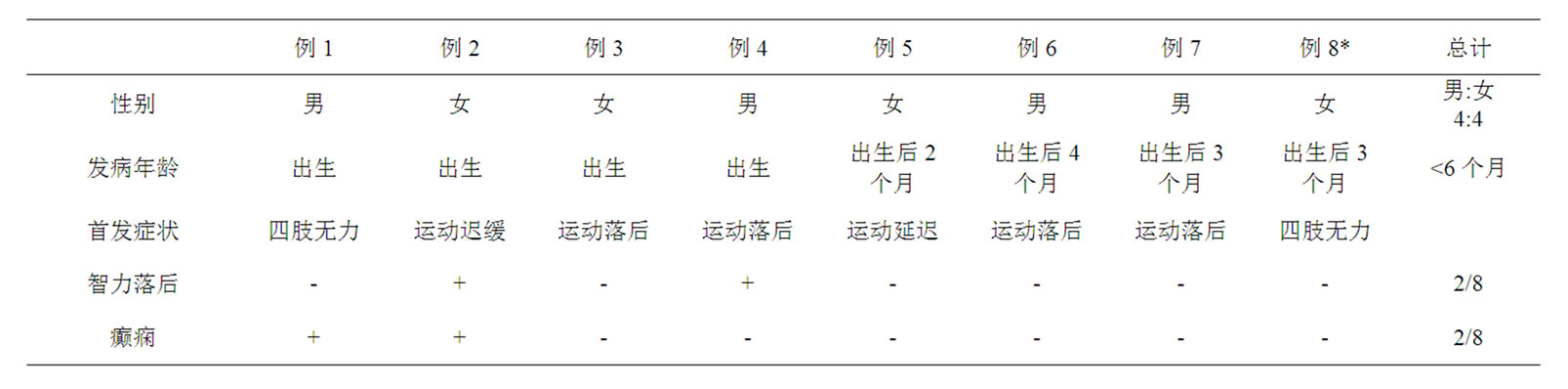
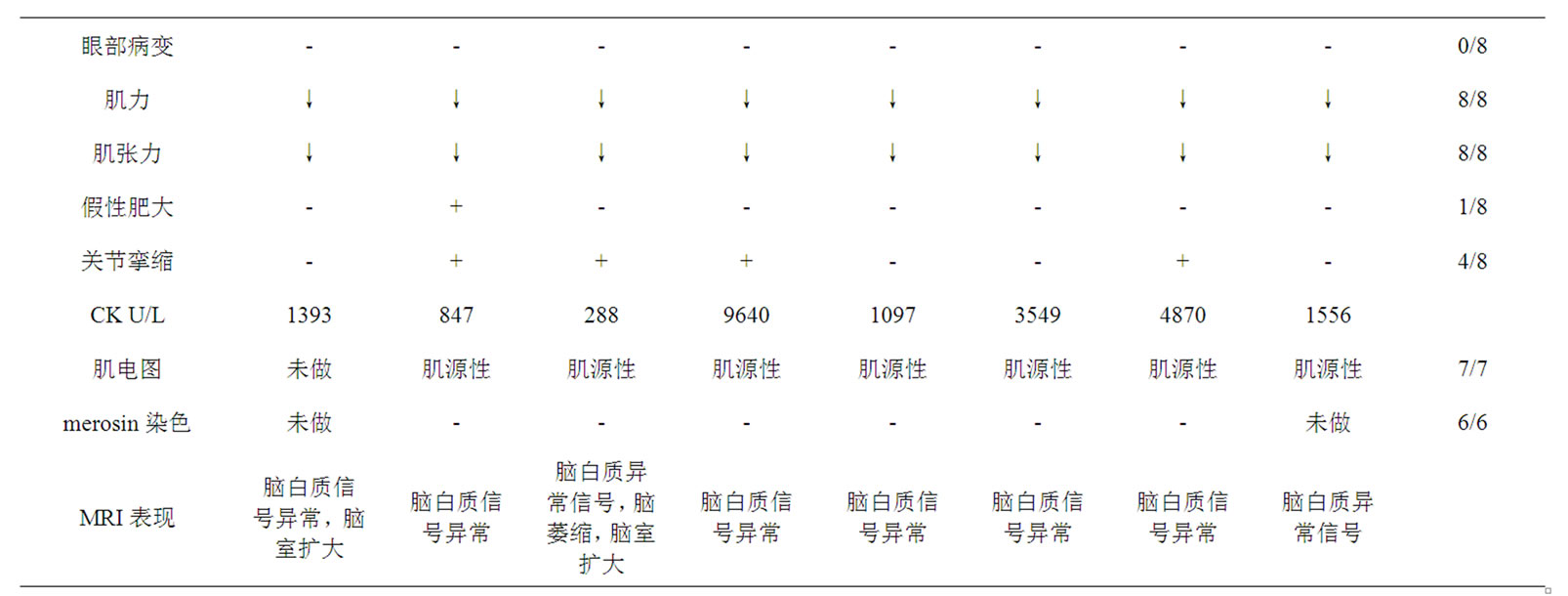
Table 1. Clinical data of 8 patients of Merosin-deficient congenital muscular dystrophy type 1A
表1. 8例Merosin缺陷性先天性肌营养不良的临床资料
不伴关节挛缩及中枢神经系统损害,肌肉病理呈肌营养不良样改变。Merosin缺陷性先天性肌营养不良在欧美多见,是先天性肌营养不良中最常见的一种类型,约占30%,而在日本仅占6%[5]。Merosin缺陷性先天性肌营养不良与Merosin缺失有关,Merosin即层粘连蛋白-α2 (Laminin-α2),是层粘连蛋白-2(Laminin-2)的3个亚单位之一(α2,β1和γ1),层粘连蛋白-2存在于横纹肌细胞外基质、周围神经、神经–肌肉接头及皮肤组织等处,层粘连蛋白(Laminin)参与细胞的粘连、分化、生长和移行。Merosin缺陷性先天性肌营养不良致病基因LAMA2等位在6q22-23[6]。Merosin缺乏可造成细胞骨架与细胞外基质的连接破坏,导致肌纤维变性、坏死,出现肌无力、肌张力低下等临床表现。
总结国内报道既往的病例和本文病例,Merosin缺陷性先天性肌营养不良患者男女性别比例无差别,起病年龄很小,8例患儿都在出生时或出生后6个月内发病,主要临床表现为运动发育里程碑落后,肌力、肌张力低下,关节挛缩,少部分患儿有语言发育迟滞和癫痫发作。但本病无近视、眼球运动障碍、视神经萎缩和视网膜变性等眼部病变,这有别于其他类型先天性肌营养不良。血清CK水平均明显升高,高于正常值5~50倍。肌电图检查显示均为肌源性损害。肌肉活检是诊断Merosin缺陷性先天性肌营养不良重要依据。肌肉病理符合肌营养不良改变,免疫组化染色提示Merosin蛋白完全消失,以此明确Merosin缺陷性先天性肌营养不良诊断。8例患儿头颅MRI均有特征性脑白质改变,主要以侧脑室旁白质病变为著,皮层下白质亦可见病变。这些脑白质病变的特点均为双侧弥漫性对称,T1低信号,T2呈高信号;与国外文献报道的Merosin缺陷性先天性肌营养不良的脑白质病变特点基本一致。目前对于Merosin缺陷性先天性肌营养不良头颅MRI的脑白质改变,其一可能因为脑血管基底膜缺乏Merosin,血管通透性增高导致脑脊液渗漏引起脑白质水肿[7];其二可能是本病脑部损害主要累及出生后1年内髓鞘化的结构,因此中枢神经系统损害可能是髓鞘发育中断的结果[8]。2例脑室系统扩大,提示脑积水。国外文献也有Merosin缺陷性先天性肌营养不良侧脑室扩大的报道。Merosin缺陷性先天性肌营养不良患儿中的小脑囊肿极少有报道,Chahnez等[9]报道3例Merosin阴性患儿均发现有小脑囊肿,此前未有报道。本研究中有2例患儿发现小脑囊肿,部分患儿可有脑干发育异常。内囊、胼胝体、基底节和丘脑均无受累。当患儿在影像学上有对称性脑白质异常表现时,结合临床表现为出生后肌力及肌张力低下、运动发育落后、实验室检查提示肌酶升高,则进一步提示Merosin缺陷性先天性肌营养不良患儿的诊断。进一步可行肌肉或皮肤组织特殊免疫组化和基因检查技术进行确诊。本科报道病例确诊是通过LAMA2基因检测最终确诊为Merosin缺陷性先天性肌营养不良。
Merosin缺陷性先天性肌营养不良需要与其他先天性肌营养不良疾病等鉴别[10]。1) α抗肌萎缩相关糖蛋白病是一种Merosin阳性先天性肌营养不良,该类疾病都有骨骼肌、脑和眼部的病变,临床症状交叉重叠,一个共同的特点是α抗肌萎缩相关糖蛋白糖基化低下。没有糖基化,α-DG就不能行使将肌肉细胞内部骨架蛋白与外部基质相连接的功能。而这些细胞外基质能够为保护肌膜不被收缩损伤提供重要的结构支持。抗肌萎缩相关糖蛋白是抗肌萎缩蛋白糖蛋白复合物(DGC)的核心,起到连接肌肉细胞和周围基质的作用,引起肌肉收缩。α抗肌萎缩相关糖蛋白病肌纤维Merosin染色呈阳性表达。先天性肌病病情多无进展,CK正常或接近正常,肌肉组织病理、酶组织化学或超微病理检查可发现诸如轴空、中央核、杆状体、管状积聚物等具有特征性的改变。2) 脊髓性肌萎缩呈进行性肌无力、萎缩,不累及面肌及眼外肌,智力正常,CK正常,肌电图呈神经元性损害,肌活检符合前角细胞病变。此外,Merosin缺陷性先天性肌营养不良MRI都有脑白质内异常信号,应与肾上腺脑白质营养不良鉴别。后者是一种主要累及中枢神经系统白质的进展性遗传性疾病,其基本特点为脑白质的髓鞘发育异常或弥漫性损害。临床可表现为四肢瘫痪、共济失调、智力低下、癫痫等,可通过酶活性测定和基因检查确诊。
综上所述,Merosin缺陷性先天性肌营养不良临床上可有肌肉、中枢和周围神经损害的肌肉疾病,诊断有赖于临床、血清CK、电生理检查、肌肉病理、头颅MRI以及基因分析。由于本病目前无特效治疗,临床上主要采取对症及运动康复为主综合疗法,有条件应尽早开展对胎儿绒毛膜滋养层细胞进行蛋白和基因产前诊断。本例患儿在传统综合治疗基础上加以免疫调节治疗,患儿运动功能逐渐进步且无倒退。因而Merosin缺陷性先天性肌营养不良发病中是否存在免疫功能紊乱,以及免疫调节对该病治疗作用等问题值得我们作更深入探索研究。
参考文献 (References)
[1] V. Allamand, P. Guicheney. Merosin-deficient congenital muscular dystrophy, autosomal recessive. European Journal of Human Genetics, 2002, 10(2): 91-94.
[2] 刘建军, 沈定国. 先天性肌营养不良合并脑白质营养不良附1例报道[J]. 罕少疾病杂志, 2010, 17(1): 50-51.
[3] 朱雯华, 赵重波, 林洁等. 先天性肌营养不良1A型的临床表现和病理改变(附1例报道)[J]. 中国临床神经科学, 2008, 16(5): 504-508.
[4] 熊晖, 姚生, 袁云等. 先天性肌营养不良的诊断及层黏连蛋白表达的意义[J]. 中华儿科杂志, 2006, 44(12): 918-923.
[5] S. Sparks, S. Quijano-Roy, A. Harper, et al. Congenital muscular dystrophy overview. Gene Reviews, 1993-2001.
[6] R. Vuolteenaho, M. Nissinen, K. Sainio, et al. Human laminin M chain (merosin): Complete primary structure, chromosomal assignment, and expression of the M and A chain in human fetal tissues. Journal of Cell Biology, 1994, 124(3): 381-394.
[7] C. C. Leite, U. C. Reed, M. C. Otaduy, et al. Congenital muscular dystrophy with merosin deficiency: 1H MR spectroscopy and diffusion-weighted MR imaging. Radiology, 2005, 235(1): 190- 196.
[8] H. J. Gilhuis, H. J. ten Donkelaar, R. B. Tanke, et al. Nonmuscular involvement in merosin-negative congenital muscular dystrophy. Pediatric Neurology, 2002, 26(1): 30-36.
[9] T. Chahnez, L. Nacim, M. Me’riam, et al. Merosin-deficient congenital muscular dystrophy with mental retardation and cerebellar cysts, unlinked to the LAMA2, FCMD, MEB and CMD1B Loci, in three tunisian patients. Neuromuscular Disorders, 2003, 13(1): 4-16.
[10] U. C. Reed. Congenital muscular dystrophy. Part I: A review of phenotypical and diagnostic aspects. Arquivos de Neuro-psiquiatria, 2009, 67(1): 144-168.