Journal of Comparative Chemistry
Vol.
03
No.
01
(
2019
), Article ID:
29350
,
10
pages
10.12677/CC.2019.31001
Progress in Molecular Docking and Molecular Dynamics Simulation
Bo Li1,2, Shuxia Liu3, Senbiao Fang3, Yubao Chen3*, Wenxiang Hu1,2,4*
1School of Chemistry and Environmental Engineering, Wuhan Institute of Technology, Wuhan Hubei
2JingdongXianghu Microwave Chemistry Union Laboratory, Beijing Excalibur Space Military Academy of Medical Sciences, Beijing
3Beijing Computing Center, Beijing
4Aerospace Systems Division, Strategic Support Troops, Chinese People’s Liberation Army, Beijing

Received: Mar. 1st, 2019; accepted: Mar. 14th, 2019; published: Mar. 21st, 2019

ABSTRACT
With the rapid development of modern life science, computational biology has gradually become one of the core disciplines and methods of modern life science research. Computational biology predicts the three-dimensional structure and dynamic characteristics of proteins from protein sequence to study the relationship between the structure and function of biological macromolecules, the interaction between biological macromolecules and ligands, and promotes the development of protein engineering, protein design and computer-aided drug design.
Keywords:Computational Biology, Molecular Docking, Molecular Dynamics, Receptor, Ligand, Molecular Design
分子对接与分子动力学计算模拟概论
李博1,2,刘书霞3,房森彪3,陈禹保3*,胡文祥1,2,4*
1武汉工程大学化学与环境工程学院,湖北 武汉
2北京神剑天军医学科学院京东祥鹄微波化学联合实验室,北京
3北京市计算中心,北京
4中国人民解放军战略支援部队航天系统部,北京
收稿日期:2019年3月1日;录用日期:2019年3月14日;发布日期:2019年3月21日

摘 要
随着现代生命科学的快速发展,计算生物学已逐步成为现代生命科学研究的核心学科和方法之一。计算生物学从蛋白质序列预测蛋白质三维结构以及动力学特征,到研究生物大分子结构与功能的关系、生物大分子之间相互作用以及生物大分子与配体的相互作用,促进了蛋白质工程、蛋白质设计和计算机辅助药物设计的发展。
关键词 :计算生物学,分子对接,分子动力学,受体,配体,分子设计

Copyright © 2019 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 引言
分子对接(Molecular docking)与分子动力学(Molecular dynamics simulation)技术是计算生物学(computational biology)中重要的一部分,在生物学研究中不断发挥着重要的作用。分子对接与分子动力学技术可以深入地阐述分子间的相互作用,并可以形象地解释相互作用的机理,特别是在药物开发中有着重要的应用,目前已经成为阐述生物学机理的重要研究方法 [1] 。分子对接与分子动力学技术还为预测生物大分子复合物结合类型与相互作用模式提供了重要的工具,并为进一步试验提供了有益的参考依据和理论支撑。在阐述生物机理与分子机制,特别是在分子甚至原子水平上对复合物结构进行预测模拟的过程中,分子对接和分子动力学技术已经成为其中最为关键且应用最为广泛的方法。鉴于两种方法的重要作用,我们在此对分子对接与分子动力学研究方法,进行初步介绍,以供参考。
2. 分子对接
2.1. 分子对接的一般原理
分子对接技术(Molecular Docking Method, MDM)是指通过电脑模拟将小分子(配体)放置于大分子靶标(受体)的结合区域,再通过计算物理化学参数预测两者的结合力(结合亲和性)和结合方式(构象),进而找到配体与受体在其活性区域相结合时能量最低构象的方法 [2] 。配体与受体结合时,彼此存在静电相互作用,氢键相互作用,范德华相互作用和疏水相互作用。配体与受体结合必须满足互相匹配原则,即配体与受体几何形状互补匹配,静电相互作用互补匹配,氢键相互作用互补匹配,疏水相互作用互补匹配(图1)。
分子对接算法主要包括以下两类:一是搜索算法(search methods),负责计算受体配体复合物的合理构象;二是打分函数(scoring function),负责评估结合亲和性以及配体位置摆放的合理性 [2] 。
2.2. 分子对接的分类
根据对接分子构象变化与否可将分子对接分为刚性对接、柔性对接和半柔性对接。刚体对接:在对接过程中,研究体系的构象不发生变化。适合考察比较大的体系,如蛋白质和蛋白质之间以及蛋白质与核酸之间的对接。半柔性对接:对接过程中,研究体系尤其是配体的构象允许在一定的范围内变化。适合处理大分子和小分子间对接,对接过程中,小分子的构象一般是可以变化的,但大分子是刚性的。柔性对接:对接过程中,研究体系的构象可以自由变化。一般用于精确计算分子间的相互作用情况,由于计算过程中体系的构象可以变化,因此计算量最大。
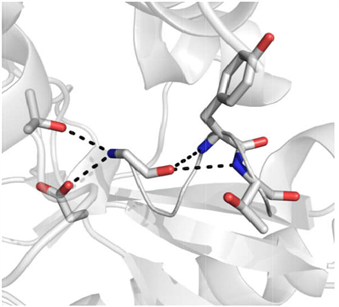 氢键作用
氢键作用
 静电相互作用
静电相互作用
 盐桥作用
盐桥作用
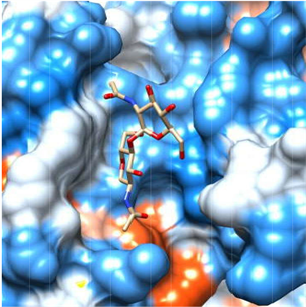 亲疏水表面
亲疏水表面
Figure 1. Schematic diagram of ligand-receptor interaction
图1. 配体与受体结合相互作用示意图
2.3. 分子对接的前期处理
小分子处理
通过经典化学构图方法对小分子化合物进行结构构建后,采用量化软件(Gaussian,ORCA等)计算分子电荷分布、分子轨道和反应活化能等对小分子进行结构优化(图2)。
蛋白质处理
对于蛋白的处理,主要是加氢、加电荷、二硫键和质子化状态方面的信息整合,其中最大的难点在于如何处理小分子周围氨基酸HIS的质子化状态,目前国际上没有一个统一的方法(图3)。

Figure 2. Schematic diagram of processing small molecules
图2. 小分子处理示意图
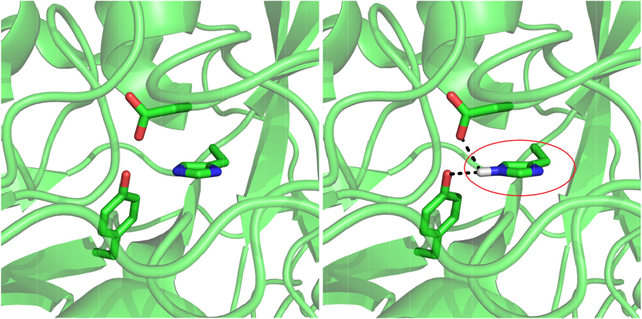
Figure 3. Schematic diagram of protein processing
图3. 蛋白质处理示意图
2.4. 分子对接示意图
分子对接其本质是两个或多个分子间的识别过程,涉及分子之间的空间匹配和能量打分,根据能量排名最终得到分子间的初步最优结构和结合模式(图4)。
“蛋白–配体”间对接软件繁杂,主要软件包括Autodock,Vina,Dock等等。
“蛋白–蛋白”与“蛋白-核酸”分子间的对接,最常用的为Rosetta,Z-DOCK等(图5)。
2.5. 分子对接的应用
近10年来,随着X-射线晶体学和高通量测序等技术的不断发展,越来越多的蛋白晶体结构得到确证,其相应的基因信息也随之公布。因此对蛋白质等生物大分子结构和功能信息的了解不断深入,产生了愈来愈多的药物靶标。随着计算科学的蓬勃发展也极大地促进了分子对接和虚拟筛选技术在药物设计领域的应用推广。新药研发是一项耗资巨大且周期漫长的系统工程,如今,计算技术己成为药物设计领域的重要手段之一,通过计算机模拟的分子对接运算,研究人员能快速准确地描述药物与靶标间的相互作用,从而缩短了药物研发周期 [2] 。
反向分子对接技术是分子对接的一个具体的新应用,它将小分子化合物作为工具,在具有三维结构的靶点数据库内进行分子对接,通过空间和能量匹配评价搜寻可能与之结合的生物大分子,进而预测理论上药物潜在的作用靶点。因此反向分子对接技术的出现为发现药物的靶点及阐明作用机制提供了一种新途径 [3] 。
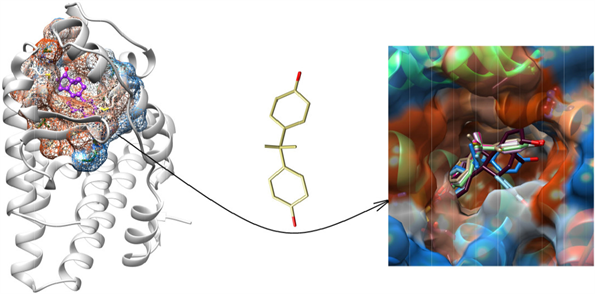

Figure 4. Analysis of docking results of protein-ligand molecules
图4. 蛋白–配体分子对接结果分析
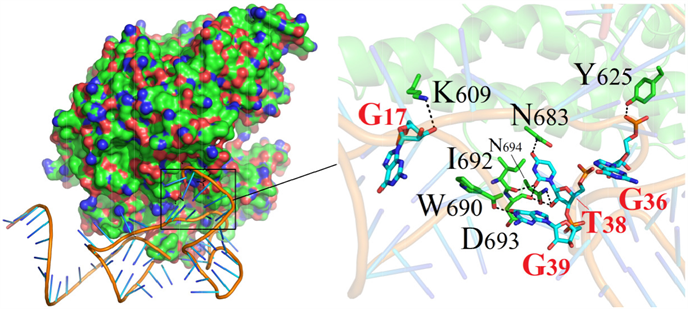
Figure 5. Molecular docking analysis of protein-nucleic acid
图5. 蛋白–核酸分子对接分析
分子对接方法在非药物发现领域中也有广泛应用 [4] [5] [6] [7] 。例如:蛋白质工程(是指按照人们的意志来改变蛋白质的结构和功能或者设计新的蛋白质的过程)、生物修复(是指使用生物体特别是微生物对环境中的污染物进行移除或者中和的过程)、生物传感器(生物大分子如抗体、受体蛋白质以及酶等,因其所具有的对底物的特异性结合能力,作为生物识别元件已广泛应用于监测环境中污染物的生物传感器中)、纳米科学(纳米材料与蛋白质相互作用)等 [8] 。
3. 分子动力学模拟计算
3.1. 分子动力学模拟概述
如果把分子对接比喻为一幅图片,那么分子动力学模拟就像是一帧桢画面组成的动态电影,丰富多彩,饶有情趣,让人情不自禁!
分子动力学(Molecular Dynamics, MD)模拟是指利用理论方法与计算技术,模拟或仿真分子运动的微观行为。分子动力学总是假定原子的运动服从某种确定的方程,这种方程可以是牛顿方程、拉格朗日方程或哈密顿方程,也就是说原子的运动和确定的轨迹联系在一起。在忽略核子的量子效应和Born-Oppenheimer绝热近似下,分子动力学的这一种假设是可行的。
分子动力学模拟广泛应用于材料科学、生物物理和药物设计等。小至单个化学分子,大到复杂生物体系或材料体系都可以用动力学模拟来研究 [9] [10] [11] 。经典MD模拟,其系统规模在一般的计算机上也可达到数万个原子,模拟时间为纳秒量级。
分子动力学方法能将分子的动态行为显示到计算机屏幕上,便于直观了解体系在一定条件下的演变过程。分子动力学方法含温度与时间,因此还可得到如材料的玻璃化转变温度、热容、晶体结晶过程、输送过程、膨胀过程、动态弛豫(relax)以及体系在外场作用下的变化过程等。
当实验研究方法不能满足研究工作的需求时,用计算机模拟却可以提供实验上尚无法得到或很难获得的重要信息;虽然计算机模拟不能完全代替实验,但为科研工作者们提供了重要的参考并且指导实验和验证某些理论假设,从而促进理论和实验的发展 [12] [13] [14] [15] [16] 。
3.2. 分子动力学模拟的基本原理
用牛顿经典力学计算许多分子在相空间中的轨迹,求解系统中的分子或原子间作用势能和系统外加约束共同作用的分子或原子的牛顿方程;模拟系统随时间推进的微观过程,通过统计方法得到系统的平衡参数或输运性质。
分子动力学的算法是有限差分方法(一种求偏微分或常微分方程和方程组定解问题的数值解的方法)。最常用的数值求解算法有Verlet算法,leap-frog算法,Beeman算法及Gear所提出的校正预测法。
1) Verlet算法
Verlet提出的Verlet算法在分子动力学中运用最为广泛,也是最简单的。它运用原子在 时刻的位置 和加速度 及 时刻的位置,计算出 时刻的位置。将粒子的位置以Taylor公式展开,即
将式中的 换为 ,得
两式相减得速度式
两式相加得
因 ,故依据上式可由 及 的位置预测 时的位置。
Verlet算法执行简明,需要的内存小,但其缺点在于速度式中含有 项,而实际计算中通常选取很
小的 值,且位置 要通过小项与非常大的两项 与 的差的相加得到,容易造成精度损失。另外,它不是一个自启动算法,新位置必须由 时刻与前一时刻 的位置得到。在t = 0时刻,只有一组位置,所以必须通过其他方法得到 的位置。获得 时刻的位置的方法之一时应用近似式 。
2) 跳蛙法(leap-frog method)
鉴于Verlet算法的一些缺点,Hockney提出了跳蛙法。跳蛙法是从Verlet法推导出来的。它在半个积分时间步长的速度,并利用这一速度计算新的位置。位置和速度表达式为
计算时假设已知 与 ,由 时的位置 计算质点所受的力与加速度 ,再预测时间为 时的速度 ,以此类推。
根据 与 ,可得时间为 时的速度为
利用跳蛙法计算仅需储存 与 两类信息,既节约储存空间,而且准确性及稳定性较高。
跳蛙法不需要计算下一步位置就可以得出速度,但需要注意的是速度并未与位置在同一时间定义,结果是动能和势能也未同时定义。所以不能直接计算总能量。
3) Beeman法
Beeman法是除Verlet跳蛙法外,另一种较为常见的方法。其积分公式如下
此方法需储存 、 与 ,储存量大于Verlet的跳蛙法。但优点在于可以引用较长的积分间隔 。Beeman方法所引用的积分步长 可为Verlet方法的3~4倍,而具有相同的准确性。
4) 校正预测法(predictor-corrector method)
Gear提出了基于预测–校正积分方法的校正预测法。因为经典运动粒子的轨迹为连续的,故于时间 时的位置、速度等可由时间 的Taylor展开式预测得
由于这些物理量来自Taylor展开式,并非由解Newton运动方程式而来,所以式中所产生的速度、加速度等并非完全正确。为了解决这个问题,用所预测的位置 计算所受的力及正确的加速度 。设正确的加速度与预测的加速度之间的误差为
得各量之校正式为
式中,c0、c1、c2、c3均为常数。这仅为Gear的一次预测校正法,也可以推展至更高次的校正法。对于大多数MD应用来说,Verlet型的算法就可以满足基本的要求了。但有时采用高阶算法更方便。一般来说Gear算法中的Taylor展开阶数越高,精度越好。但占用内存比较大,这对于大量分子系统的模拟不如Verlet算法方便。当 减小时,Gear的高次预测校正法误差较小,但 增大时,Verlet方法较佳。
3.3. 分子动力学模拟中的重要概念
1) 周期性边界条件:只模拟实际物质中很小一部分,使模拟体系成为无限的具有相同性质的分子体系的,该部分在三维空间中周期性的存在,代表整个体系。
2) 势函数:势函数是描述原子(分子)间相互作用的函数。原子间的相互作用控制着原子间的相互作用行为,从根本上决定材料的所有性质,这种作用具体由势函数来描述,在分子动力学模拟中,势函数的选取对模拟的结果起着决定性的作用。
3) 系综:在一定的宏观条件下,大量性质和结构完全相同的、处于各种运动状态的、各自独立的系统的集合。系综是用统计方法描述热力学系统的统计规律性时引入的一个基本概念。
正则系综是组成系综的系统是由N个粒子组成的,同温度为T的很大的热源相接触并达到热平衡。也可以这样设想:取大数M个体积为V、粒子数为N的相同的系统构成系综,其中任意一个系统均可作为被研究的系统,其余 个系统起着恒温槽的作用,系统间有能量交换,并共同处于热平衡。
微正则系综是指系统原子数N、体积V、能量E保持不变,又称为NVE系综。
等温等压系综是正则系综的推广,是统计力学系综的一种。这个系综是具有恒定温度和压强的体系。每个系综内的体系可以和其他体系进行能量和体积交换。但系综内各体系的能量总和以及体积总和是固定的,而且各体系有相同的粒子数。
4) 积分步长:分子动力学计算的基本思想是赋予分子体系初始运动状态之后利用分子的自然运动在相空间中抽取样本进行统计计算,积分步长就是抽样的间隔。选取合适的积分步长的原则是积分步长小于系统中最快运动周期的十分之一。在节省时间的同时也保证计算的精确性。
3.4. 分子动力学模拟的应用
近年来,分子动力学模拟的应用范围越来越广泛。例如:我们采用分子对接和分子动力学(MD)模拟方法研究了芬太尼类化合物与阿片μ受体的相互作用机制。先用AutoDock4.0程序将芬太尼类化合物对接到同源模建的阿片μ受体结构中,再用GROMACS程序包在水溶液体系中分别对12个芬太尼激动剂和阿片μ受体蛋白复合物进行了MD模拟研究,优化对接复合物的结构,最后利用MM-PBSA方法,在APBS程序中计算芬太尼类衍生物与阿片μ受体的结合自由能,计算出的受体配合物结合常数(Ki)与其实验值吻合较好,并预测了化合物的活性排序 [17] 。
陈莹等运用分子动力学方法模拟了小尺寸金属团簇的熔化过程 [18] 。汪志刚等采用分子动力学结合嵌入原子多体势,模拟了不同半径的Ni纳米团簇的升温熔化过程 [19] 。郑燕升等利用分子动力学模拟和量子化学计算方法对不同离子液体进行研究,获得离子液体的结构性质、光谱性质( 红外光谱、拉曼光谱)及离子液体催化反应机理 [20] 。王丽等采用常温、常压分子动力学模拟技术,研究了在周期性边界条件下,由864个Cu原子构成的模型系统的熔化、结晶过程 [21] 。
4. 结语
分子对接及分子动力学已成为计算生物学必要手段,在生物学、医学、药物学、农学和环境科学等领域都发挥着重要的作用。分子对接及分子动力学是计算生物学的重要组成部分,其发展依赖于各个交叉学科的发展和渗透,同时各个交叉学科的迅速发展也给计算生物学的发展创造了有利条件。
随着人类基因组研究和蛋白质组研究的不断深入,分子对接及分子动力学技术在未来将面临更加巨大的挑战,特别是人工智能技术和计算资源的巨大需求,将不断推动这两项技术的不断前进和变革,在揭示生命奥秘中也将会更加趋于成熟和完善。分子对接及分子动力学技术对生物学发展的贡献,也将促进新的交叉学科继续产生,共同推动生命科学进入一个全新的境界 [22] [23] [24] 。
分子对接与分子动力学技术,为预测生物大分子复合物结合类型与相互作用模式提供了重要的工具,并为进一步试验提供了有益的参考依据和理论支撑。在阐述生物机理与分子机制,特别是在分子甚至原子水平上对复合物结构进行预测模拟的过程中,分子对接和分子动力学技术已经成为其中最为关键且应用最为广泛的方法。随着生物学数据量的不断增长,分子对接和分子动力学技术的重要性和复杂性逐渐彰显,面临的生物学问题越复杂,越需要这两种技术的参与和应用。计算生物学,特别是分子对接与分子动力学技术正在成为现代生物学研究的核心技术,成为当今生命科学最具活力的新兴科研工具 [25] [26] [27] 。
文章引用
李 博,刘书霞,房森彪,陈禹保,胡文祥. 分子对接与分子动力学计算模拟概论
Progress in Molecular Docking and Molecular Dynamics Simulation[J]. 比较化学, 2019, 03(01): 1-10. https://doi.org/10.12677/CC.2019.31001
参考文献
- 1. 徐书华, 金力. 计算生物学[J]. 科学, 2009, 61(4): 34-37.
- 2. 赵晨, 夏春光, 于敏, 潘月, 王辂. 分子对接软件在药物设计中的应用[J]. 中国抗生素杂志, 2015, 40(3): 234-240.
- 3. 任洁, 魏静. 分子对接技术在中药研究中的应用[J]. 中国中医药信息杂志, 2014, 21(1): 123-125.
- 4. Lu, J. (2011) Molecular Docking and Molecular Dynamics Simulations of Inhibitors Binding to Jack Bean Urease. Acta Chimica Sinica, 69, 2427-2433.
- 5. Shoichet, B., Kuntz, I. and Bodian, D. (1992) Molecular Docking Using Shape Descriptors. Journal of Computational Chemistry, 13, 380-397. https://doi.org/10.1002/jcc.540130311
- 6. Doman, T., McGovern, S., Witherbee, B., et al. (2002) Molecular Docking and High-Throughput Screening for Novel Inhibitors of Protein Tyrosine Phosphatase-1B. Journal of Medicinal Chemistry, 45, 2213-2221. https://doi.org/10.1021/jm010548w
- 7. Li, Z., et al. (2011) Molecular Docking Methods Based on Drug Dis-covery Grid. Journal of Huazhong University of Science and Technology, 32, 259-261.
- 8. 许先进, 王存新. 分子对接方法在药物发现之外领域的应用[J]. 北京工业大学学报, 2017, 43(12): 1872-1880.
- 9. Gao, W. (2015) Mo-lecular Dynamics Simulation of Fracture in Micro and Nano Scale. Foreign Electronic Measurement Technology, 10, 178-190.
- 10. Sun, X., Pei, S., Wang, J., et al. (2017) Coarse-Grained Molecular Dynamics Simulation Study on Spherical and Tube-Like Vesicles Formed by Amphiphilic Copolymers. Journal of Polymer Science Part B: Polymer Physics, 55, 1220-1226. https://doi.org/10.1002/polb.24376
- 11. Smith, J., Borodin, O., Smith, G. and Kober, E.M. (2007) A Molecular Dynamics Simulation and Quantum Chemistry Study of Poly(Dimethylsiloxane)-Silica Nanoparticle Interactions. Journal of Polymer Science Part B: Polymer Physics, 45, 1599-1615. https://doi.org/10.1002/polb.21119
- 12. 李春艳, 刘华, 刘波涛. 分子动力学模拟基本原理及研究进展[J]. 广州化工, 2011, 39(4): 11-13.
- 13. Frank, M., Gutbrod, P., Hassayoun, C. and von der Lieth, C.-W. (2003) Dynamic Molecules: Molecular Dynamics for Everyone. An Internet-Based Access to Molecular Dynamic Simulations: Basic Concepts. Journal of Molecular Modeling, 9, 308-315. https://doi.org/10.1007/s00894-003-0144-y
- 14. Yoshikawa, K. (2005) Single Macromolecules: Hierarchic Thermodaynamics, Irreversibility and Biological Function. Journal of Biological Physics, 31, 243-248. https://doi.org/10.1007/s10867-005-7285-4
- 15. Xu, Y. (2014) Molecular Dynamics Simulation of Water Mole-cules Adsorbed at Muscovite Surface. CIESE Journal, 12, 226-239.
- 16. Markevitch, A., Smith, S., Romanoy, D., et al. (2003) Nonadiabatic Dynamics of Polyatomic Molecules and Ionsin Strong Laser Fields. Physical Review A, 68, Article ID: 011402. https://doi.org/10.1103/PhysRevA.68.011402
- 17. 李博, 刘明, 胡文祥. 芬太尼类化合物与阿片μ受体相互作用的分子对接与分子动力学模拟[J]. 物理化学学报, 2010, 26(1): 206-214.
- 18. 陈莹, 王秀英, 赵俊卿. 小尺寸金属团簇熔化过程的分子动力学模拟[J]. 物理化学学报, 2008, 24(11): 2042-2046.
- 19. 汪志刚, 邱姝颖, 文玉华. 纳米团簇熔化过程的分子动力学模拟[J]. 原子与分子物理学报, 2008, 25(4): 848-852.
- 20. 郑燕升, 卓志昊, 莫倩, 李军生. 离子液体的分子模拟与量化计算[J]. 化学进展, 2011, 23(9): 1862-1870.
- 21. 王丽, 边秀房, 李辉. 金属Cu 熔化结晶过程的分子动力学模拟[J]. 化学物理学报, 2000, 13(5): 544-550.
- 22. 程妍, 刘仲林. 计算生物学——一门充满活力的新兴交叉学科[J]. 科学学与科学技术管理, 2006(3): 11-15.
- 23. 梁丹, 薄文浩, 姜立波. 生物学新兴前沿学科——计算生物学[J].中国林业教育, 2007, 35(1): 139-142.
- 24. Liu, M. and Hu, W.X. (2017) Using Deep Belief Network and Computation Methods to Improve Opioid Receptor Biological Activity Prediction, Novel Agonists and Antagonists Structural Modeling. 2nd International Conference on Manufacturing Science and Information Engineering (ICMSIE), Guangzhou, 538-543.
- 25. Wang, L., Hu, W.X. and Liu, X.L. (2009) The Modeling of Three-Dimensional Structure of Human μ-Opioid Receptor and the Study of Molecular Docking of Fentanyl Analogs. Computers and Applied Chemistry, 26, 746-750.
- 26. 胡文祥, 李博. 比较化学——构筑量子化学通向分子药学的桥梁[M]. 北京: 化学工业出版社, 2013.
- 27. 刘明, 马密霞, 韩谢. 《书评: 比较化学——构筑量子化学通向分子药学的桥梁》[J]. 比较化学, 2018, 2(1): 6-10.
NOTES
*通讯作者。