Applied Physics
Vol.06 No.05(2016), Article ID:17514,9
pages
10.12677/APP.2016.65013
First-Principle Study of Doping Atoms Effects on Structural Stability and Electronic Properties of α-Cr2O3 Oxide
Shengyi Zhuang1,2, Zhuxia Zhang1,2*, Hui Huang1,2, Jian Wang1,2, Peide Han1,2*
1Key Laboratory of Interface Science and Engineering in Advanced Materials, Taiyuan University of Technology, Ministry of Education, Taiyuan Shanxi
2College of Materials Science and Engineering, Taiyuan University of Technology, Taiyuan Shanxi

Received: Apr. 21st, 2016; accepted: May 4th, 2016; published: May 10th, 2016
Copyright © 2016 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



ABSTRACT
The structural stability and electronic property of α-Cr1.5M0.5O3 with doping atoms are studied by first-principle calculations. The cohesive energy of α-Cr2O3 with Fe, Mo, Nb, Ni, Mn, Al and Si elements indicates that Nb, Al, Mo and Mn atoms are effective for improving the binding ability of α-Cr2O3, especially Nb in α-Cr2O3 oxide. Gibbs free energy proves that these doping atoms’ solid solution in α-Cr2O3 makes the structure more stable at the temperature range of 200 - 1000 K, especially Ni and Si in α-Cr2O3 oxide. Compared with the band gap width of α-Cr1.5M0.5O3 (M = Cr, Fe, Mo, Nb, Ni, Mn, Al, Si), Mo and Al are elements which are effective for increasing band gap width of α-Cr1.5M0.5O3, namely Mo and Al can make the structure more electrochemically stable.
Keywords:Cr2O3, Density Functional Theory, Cohesive Energy, Electronic Property, Doped

掺杂原子对α-Cr2O3结构稳定性和电子特性的第一性原理研究
庄晟逸1,2,张竹霞1,2*,黄慧1,2,王剑1,2,韩培德1,2*
1太原理工大学,教育部新材料界面科学与工程教育部重点实验室,山西 太原
2太原理工大学材料科学与工程学院,山西 太原

收稿日期:2016年4月21日;录用日期:2016年5月4日;发布日期:2016年5月10日

摘 要
本文采用第一性原理研究了掺杂原子对α-Cr2O3结构稳定性和电子特性的影响。研究结果表明Fe、Mo、Nb、Ni、Mn、Al、Si掺杂于α-Cr2O3复合体系的结合能均为负值,这些元素构成的α-Cr2O3复合氧化物均具有稳定的结构,尤其Nb的作用最为明显。吉布斯自由能来看,掺杂原子后α-Cr2O3复合体系结构稳定性在200~1000 K温度范围内,随着温度的升高结构稳定性逐步增强,尤以Ni和Si最为明显。由布居数和态密度分析了复合体系α-Cr1.5M0.5O3的电化学活性,计算结果来看Mo和Al能使α-Cr2O3构成的复合氧化物的带隙宽度稍微增加,而Si和Mn则使α-Cr2O3的带隙宽度变窄,即Mo和Al能提高α-Cr1.5Mo0.5O3和α-Cr1.5Mo0.5O3复合氧化物的电子化学稳定性。
关键词 :三氧化二铬,密度泛函理论,结合能,电子特性,掺杂

1. 引言
Cr2O3是一种重要的陶瓷材料,由于它具有高的化学稳定性,强的耐磨性以及低的热传导性,作为涂层或耐火材料常用于各种恶劣的工况环境下 [1] 。同时高Cr合金如不锈钢以其表面形成致密的Cr2O3保护膜而展现出很强的耐蚀性,从而保护基体不被外界腐蚀介质侵蚀 [2] 。
最近,将不同的金属元素诸如Ca、Li、Al和Fe摻杂于Cr2O3形成复合氧化物的结构与电子特性已有了较多的理论研究 [3] - [6] 。如Ca摻杂于Cr2O3中,通过对带隙结构的调控,表现出铁磁性 [3] ;Li掺杂到Cr2O3中可以用作高温氧化条件下的导电陶瓷材料等 [4] ,Cr2O3作为涂层已广泛应用于高温氧化环境,刚玉型的CrxAl2−xO3固溶体具有比α-Cr2O3及α-Al2O3更为稳定的结构 [5] ,大量的研究报道主要是为了探究α-Cr2O3形成的复合物在催化材料和防腐涂层上的应用。而对于表面形成抗氧化性极高的致密Cr2O3氧化层的高铬合金,Cr2O3与高铬合金中金属原子构建成的复合氧化物的理论研究却很少。已有研究表明高铬合金中添加少量的合金化元素,如Ni,Nb,Al,Mo,Ce等,通常能使高铬合金在高温氧化环境下的抗腐氧化性能得到明显改善 [7] 。鉴于实验上研究α-Cr2O3复合氧化物的难度,而相关合金元素对α-Cr2O3复合氧化物结构和性能影响机理尚不十分清楚,因此亟待开展相关α-Cr2O3复合氧化物结构稳定性和电子特性原子层次的理论研究。
到目前为止,针对Cr2O3的实验研究已有很多 [8] [9] ,但是针对性开展金属原子固溶于Cr2O3形成复合氧化物的实验和理论研究却较少。据我们所知,目前还没有针对Fe、Mo、Nb、Ni、Mn、Al和Si与α-Cr2O3形成α-Cr2O3复合氧化物的结构稳定性和电化学性能上的理论研究,本研究将采用密度泛函理论系统分析这些过渡金属元素对三元复合氧化物α-Cr1.5M0.5O3的结合能力和电子特性的影响。
2. 计算方法
本文采用基于第一性原理平面赝势波的CASTEP (Cambridge Sequential Total Energy Package)模块。选取含有10个原子的斜六面体α-Cr2O3单胞为研究对象。α-Cr2O3为 的刚玉型结构,以此结构为基础,我们选取Fe,Mo,Nb,Ni,Mn,Al和Si等七种不同的金属原子分别取代α-Cr2O3中的一个Cr原子,构成摻杂金属原子的α-Cr2O3复合体系。如图1为掺杂金属原子的α-Cr2O3复合体系的原子结构模型。由于α-Cr2O3属于过渡金属氧化物,具有很强的电子相关性,是介于电荷转移绝缘体和莫特-哈伯德绝缘体之间的中间型绝缘体 [10] 。基于DFT-GGA的方法忽略了过渡金属元素d轨道电子之间的很强的库伦斥力,为此本文采用考虑原子内强电子相关作用的非抑制性从头计算(UHF)近似方法,即DFT + U方法 [3] 。DFT + U方法能够对氧化物的结构,电子特以及磁性特征等给出较为精准的结果。计算中U值为4 eV,屈服值为每个单位为0.179 eV。计算得到的α-Cr2O3的带隙值为3.01 eV,与实验值3.3 eV十分接近 [11] 。
的刚玉型结构,以此结构为基础,我们选取Fe,Mo,Nb,Ni,Mn,Al和Si等七种不同的金属原子分别取代α-Cr2O3中的一个Cr原子,构成摻杂金属原子的α-Cr2O3复合体系。如图1为掺杂金属原子的α-Cr2O3复合体系的原子结构模型。由于α-Cr2O3属于过渡金属氧化物,具有很强的电子相关性,是介于电荷转移绝缘体和莫特-哈伯德绝缘体之间的中间型绝缘体 [10] 。基于DFT-GGA的方法忽略了过渡金属元素d轨道电子之间的很强的库伦斥力,为此本文采用考虑原子内强电子相关作用的非抑制性从头计算(UHF)近似方法,即DFT + U方法 [3] 。DFT + U方法能够对氧化物的结构,电子特以及磁性特征等给出较为精准的结果。计算中U值为4 eV,屈服值为每个单位为0.179 eV。计算得到的α-Cr2O3的带隙值为3.01 eV,与实验值3.3 eV十分接近 [11] 。
吉布斯自由能的计算,采用的是基于密度泛函理论的DMol3模块来完成的。参数设置同样是广义梯度近似(GGA)的方法,平面波截断能为500 eV,采用6 × 6 × 6的Monkhorst-Pack k点网格对布里渊区进行划分,优化过程中,对原子的占位及晶胞的晶格常数进行充分驰豫。我们优化后α-Cr2O3的晶胞参数为a = 5.647 (Å),V = 93.281 (Å3),与文献 [12] 的计算结果a = 5.40 (Å),V = 97.5 (Å3)非常接近,且与实验值 [13] a = 5.359 (Å),V = 96.50 (Å3)之间的误差低于5.37%,表明本计算方法是可行的,后续计算结果均是在这些参数下获得的。
3. 结果与讨论
3.1. 结构稳定性
首先对α-Cr1.5M0.5O3 (M = Fe,Mo,Nb,Ni,Mn,Al和Si)复合氧化物进行了结构优化,表1列出了所有复合物α-Cr1.5M0.5O3 (M = Cr,Fe,Mo,Nb,Ni,Mn,Al和Si)优化后的结构参数。可以看到,这六种元素对单胞常数的影响是不同的,Fe、Ni、Al和Si使得复合氧化物的单胞常数减小,而Mo和Nb使得单胞常数增加。而对体积的影响来看,除Si使得减小外,其余元素都使得体积增加。
为了分析复合氧化物α-Cr1.5M0.5O3的结构稳定性,我们计算了复合氧化物的形成能 [14] 和结合能 [15] ,计算公式如下:
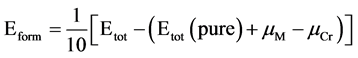 (1)
(1)
 (2)
(2)
公式(1)表示的是不同的金属原子M固溶到α-Cr1.5M0.5O3中,所形成的形成能。式中Etot(Cr1.5M0.5O3),Etot(Cr2O3)分别表示复合氧化物和纯α-Cr2O3的总能。由于本研究中置换原子的个数为一,μM表示单个金属原子M (M = Cr,Fe,Mo,Nb,Ni,Mn,Al和Si)的化学势。在T = 0 K和P = 0 Pa时,形成焓的值等于形成能的值,这里忽略零点振动能。形成能/焓可以用来分析化合物的形成能力,这里可确定含掺杂原子α-Cr1.5M0.5O3复合氧化物的形成能力。公式(2)给出了复合氧化物的结合能,其中Etot(Cr1.5M0.5O3)为总能,同公式(1),EM,ECr,EO分别表示M,Cr和O原子在晶胞中的单个原子能,所有能量的结果均是在各组分的基态结构条件下获得。结合能的大小可确定复合氧化物的结合能力。
图2即为复合氧化物α-Cr1.5M0.5O3 (M = Cr,Fe,Mo,Nb,Ni,Mn,Al和Si)的形成能。从图中可以看出,七种元素对复合氧化物形成能的影响各不相同,其中α-Cr1.5Nb0.5O3,α-Cr1.5Al0.5O3,α-Cr1.5Mo0.5O3 和α-Cr1.5Mn0.5O3的形成能为负值,表明Nb、Al、Mo和Mn极易溶于α-Cr2O3形成复合氧化物,而Ni、Si不易溶于α-Cr2O3形成α-Cr1.5Ni0.5O3和α-Cr1.5Si0.5O3。为了进一步了解所构成的复合氧化物的结构稳定性,我们也计算了它们的结合能。

Figure 1. Primitive cell for corundum-type oxide, α-Cr1.5M0.5O3 (M = Cr, Fe, Mo, Nb, Ni, Mn, Al and Si)
图1. 含掺杂金属α-Cr1.5M0.5O3 (M = Cr,Fe,Mo,Nb,Ni,Mn,Al和Si)复合结构原子模型
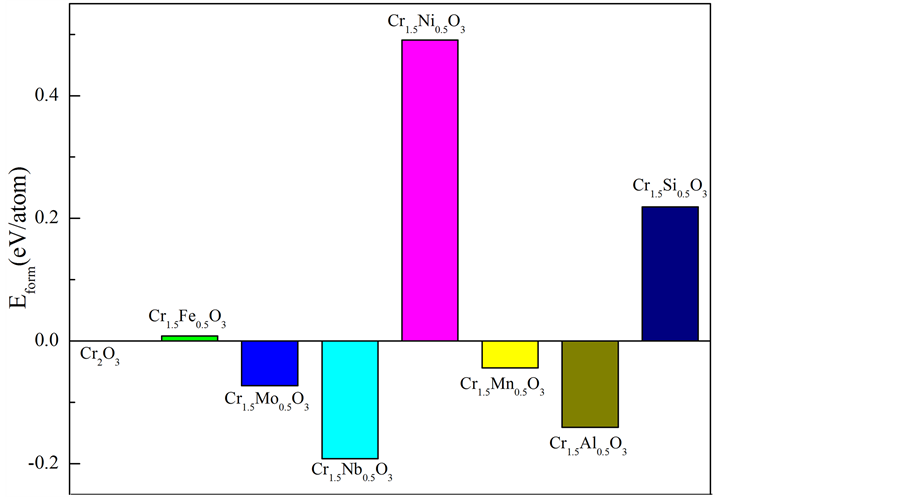
Figure 2. Formation energy for α-Cr1.5M0.5O3, (M = Cr, Fe, Mo, Nb, Ni, Mn, Al and Si)
图2. α-Cr1.5M0.5O3 (M = Cr,Fe,Mo,Nb,Ni,Mn,Al和Si)的形成能

Table 1. Main results obtained for ground state of α-Cr1.5M0.5O3 (M = Cr, Fe, Mo, Nb, Ni, Mn, Al and Si): lattice constants a, optimum volumes V
表1. 优化后α-Cr1.5M0.5O3 (M = Cr,Fe,Mo,Nb,Ni,Mn,Al和Si)的晶胞参数
结合能可以反映出体系的结构稳定性,图3给出了α-Cr1.5M0.5O3 (M = Cr,Fe,Mo,Nb,Ni,Mn,Al和Si)的结合能。掺杂元素分别为Fe,Mo,Nb,Ni,Mn,Al和Si时,复合氧化物的结合能均为负值,表明这些复合物均为热力学稳定的结构。其中Mo、Nb、Mn、Al所形成复合氧化物的结合能低于α-Cr2O3,具有更高的结构稳定性。
总体来看,Nb、Al、Mo和Mn四种原子易于溶于α-Cr2O3中形成复合氧化物,同时所形成的复合氧化物又具有较高的结构稳定性。由于以上计算结果对应于0 K,这些复合化合物往往服役于高温环境下,为此以下我们将计算这些复合氧化物在不同时的吉布斯自由能。

Figure 3. Cohesive energy for Cr1.5M0.5O3 (M = Cr, Fe, Mo, Nb, Ni, Mn, Al and Si)
图3. α-Cr1.5M0.5O3 (M = Cr,Fe,Mo,Nb,Ni,Mn,Al和Si)的结合能
3.2. 不同温度下的吉布斯自由能
材料的热力学特性可以用来描述体系结构的稳定性,通常吉布斯自由能越低,化合物体系的热力学稳定性越高。图4给出了α-Cr2O3及七种复合氧化物体系在200 K~1000 K之间的吉布斯自由能变化曲线。图中可以看出,金属原子Fe、Mo、Nb、Ni、Mn、Al和Si固溶到α-Cr2O3中形成的复合氧化物,随着温度的升高,吉布斯自由能均呈现下降的趋势。即在200 K~1000 K温度范围内,温度越高,α-Cr2O3及七种复合氧化物的稳定性越高,具体来看由Fe,Mo,Nb,Ni,Mn,Al和Si形成的复合体系的吉布斯自由能要低于纯的α-Cr2O3,因而这几种合金元素固溶到α-Cr2O3中,可以使结构更加稳定。对比在同一温度下,吉布斯自由能的由小到大的变化顺序为:α-Cr1.5Si0.5O3、α-Cr1.5Ni0.5O3、α-Cr1.5Al0.5O3、α-Cr1.5Nb0.5O3、α-Cr1.5Fe0.5O3、α-Cr1.5Mn0.5O3、α-Cr1.5Mo0.5O3和α-Cr2O3。总体来讲,同一温度下掺杂原子形成复合物的吉布斯自由能的值变化并不是很大,当温度升高到490 K时,Si和Ni原子比Mo、Nb、Al更容易固溶到α-Cr2O3中,而超过1000 K时,合金元素Ni、Si比Mo、Mn、Fe更容易固溶到α-Cr2O3中。
3.3. 复合体系的电子特性
通过α-Cr1.5M0.5O3的电子结构,可以更好的理解掺杂原子的结构稳定性和电子特性。图5给出了α-Cr1.5M0.5O3 (M = Cr,Fe,Mo,Nb,Ni,Mn,Al和Si)复合氧化物的态密度图,对比来看,纯α-Cr2O3导带与价带之间有明显的带隙,价电子主要集中在−8.16 eV到18.82 eV之间,主要由Cr的3d轨道和O的2p轨道共同作用的结果,以上态密度曲线与文献 [6] 的计算结果相符。图5(a)中可以看出,α-Cr2O3的态密度可以分成明显的两部分,为了了解摻杂原子对复合物电子性能的影响,对比了纯α-Cr2O3与α-Cr1.5M0.5O3复合物的态密度。图5(b)可以看出α-Cr1.5Al0.5O3相较于α-Cr2O3的态密度,一小部分的价带发生了右移,并且导带底也向左发生了轻微的移动。由于Al原子的加入,价带和导带的电子数都有一定的增加,但大部分的电子主要集中在低能级,并且费米能级处电子数仍为零。图5(c)中α-Cr1.5Mo0.5O3的价带和导带均左移,并且电子数减少,同时因为Mo原子的加入使得导带的一部分空带被占据。相较于

Figure 4. Temperature-dependent Gibbs free energy for α-Cr1.5M0.5O3 (M = Cr, Fe, Mo, Nb, Ni, Mn, Al and Si)
图4. α-Cr1.5M0.5O3 (M = Cr,Fe,Mo,Nb,Ni,Mn,Al和Si)复合体系吉布斯自由能


Figure 5. Density of state for α-Cr1.5M0.5O3 (M= Cr, Fe, Mo, Nb, Mn and Al). The dotted line marks the Fermi level (Ef)
图5. α-Cr
上述两种复合体系,图5(d)中Nb的加入导带左移程度更明显,使得α-Cr1.5Nb0.5O3复合物的带隙变窄,同时导带处一部分空带被填满,电子数减少。图5(e)的α-Cr1.5Mn0.5O3复合体系,由于Mn原子的加入,导带明显的左移,价带右移,结果使得带隙明显变窄,且费米能级处有活泼电子,导带底部价电子数显著增加。图5(f)代表的是α-Cr1.5Fe0.5O3体系的态密度,导带左移的量不大,但是导带底出现一个明显的峰,说明处于这个能级的电子数剧增,这主要是由Fe的3d轨道贡献的。
下面通过布居分析,计算掺杂原子临近区域O原子的电子转移情况。表2列出了纯α-Cr2O3以及复合氧化物中摻杂原子与它们最近邻的O原子的电子转移数的相对值。电子转移数的减少意味着自由电子数增加,一般合金元素将失去电子,进而使其周围的O原子电子数增加。对于α-Cr1.5M0.5O3体系来看,掺杂原子电子转移数的变化均较小,只有Fe和Ni原子获得了电子,与它临近的O原子失去电子,这就会引起它周围离子键的减弱,进而使体系的形成焓增加,使体系结构稳定性变差。相较而言,Nb、Al以及Mo原子失去电子,这使得它构成的离子键增强,形成焓降低,使得体系结构稳定性增强。
只有费米能级处最外层的电子,能够挣脱原子核的束缚变成自由电子,自由电子由价带跃迁到导带,需要克服一个很大的能垒,这个能垒的大小即带隙宽度。其取决于价带与导带之间的能量差。为了更好的研究复合体系的电化学特性,我们绘制了所有复合体系的带隙结构(如图6)。随着能量差的增大,复合体系的电化学活性降低,即体系结构稳定性增强。已知α-Cr2O3是宽带隙的半导体。当Mo原子取代一个Cr原子,固溶到基体中后,体系的带隙变宽,也就是价带与导带之间的能垒变大,致使自由电子发生跃迁的难度增大,体系的电化学活性降低。同时对于Al来说,能量差虽然减少,但是减少的值非常小,体
Table 2. Bader charges of doping atoms and its nearest O atoms
表2. 掺杂原子与最近邻的O原子的布居数
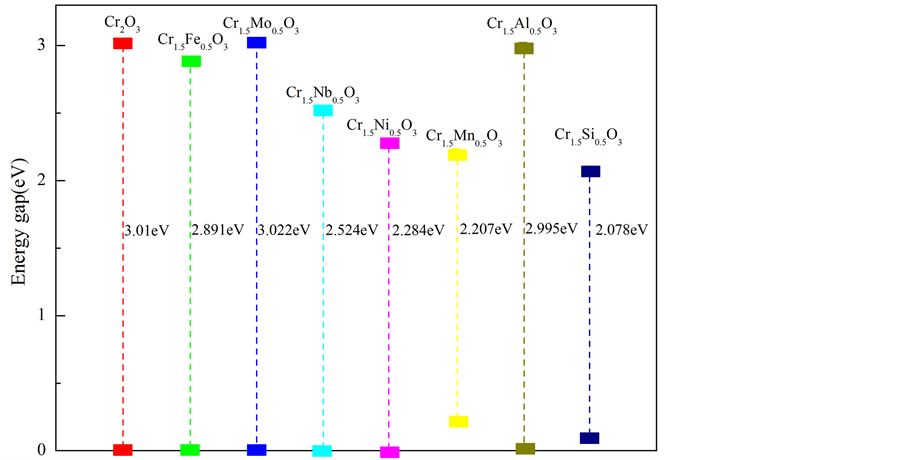
Figure 6. A sketch map of band-gap width for Cr1.5M0.5O3 (M = Cr, Fe, Mo, Nb, Ni, Mn, Al and Si)
图6. α-Cr1.5M0.5O3 (M = Cr,Fe,Mo,Nb,Mn和Al)复合氧化物的带隙示意图
系的电化学活性没有太大的变化。相较于其他的合金元素来说,α-Cr1.5Mo0.5O3、α-Cr1.5Al0.5O3复合氧化物体系的电化学活性较低,即较其他合金元素构成的复合体系具有更好的结构稳定性。同时,α-Cr1.5Si0.5O3和α-Cr1.5Mn0.5O3体系的能量差非常小,甚至价带和费米能级相交,致使费米能级处自由电子数增多,这些电子很容易跃迁到能量更高的导带,致使整个体系结构稳定性变差。
4. 结论
本文系统分析了含有Fe,Mo,Nb,Ni,Mn,Al和Si原子,形成α-Cr2O3的复合氧化物的结构稳定性与电化学活性,得到以下几点结论。
1) 形成能和结合能的计算表明,Nb,Al,Mo,Mn均极容易溶于α-Cr2O3形成复合氧化物,并且所构成复合体系具有较好的结构稳定性。
2) 在200~1000 K温度范围间,掺杂原子均可稳定存在于α-Cr2O3中,且随着温度的升高,Ni和Si比Mo和Al更容易固溶到α-Cr2O3中,形成结构稳定的复合物。
3) 态密度和能带结构的分析表明,Mo和Al能够降低α-Cr1.5M0.5O3复合体系电化学活性,也即增加复合体系的结构稳定性。
致谢
感谢国家自然科学基金项目(51371123)、山西省自然科学基金项目(2014011002)的支持。
文章引用
庄晟逸,张竹霞,黄 慧,王 剑,韩培德. 掺杂原子对α-Cr2O3结构稳定性和电子特性的第一性原理研究
First-Principle Study of Doping Atoms Effects on Structural Stability and Electronic Properties of α-Cr2O3 Oxide[J]. 应用物理, 2016, 06(05): 91-99. http://dx.doi.org/10.12677/APP.2016.65013
参考文献 (References)
- 1. Zhu, L., Peng, X., Yan, J. and Wang, F. (2004) Oxidation of a Novel Chromium Coating with CeO2 Dispersions. Oxidation of Metals, 62, 411–426. http://dx.doi.org/10.1007/s11085-004-0921-8
- 2. Marcus, P., Ed. (2002) Corrosion Mechanisms in Theory and Practice. 2nd Edition, Marcel Dekker, New York. http://dx.doi.org/10.1201/9780203909188
- 3. Maldonado, F., Rivera, R. and Stashans, A. (2012) Structure, Electronic and Magnetic Properties of Ca-Doped Chromium Oxide Stu Died by the DFT Method. Physica B, 407, 1262-1267. http://dx.doi.org/10.1016/j.physb.2012.01.116
- 4. Crosbie, G.M., Tennenhouse, G.J., et al. (1984) Electronically Conductive Magnesia Doped Oxide Ceramics for Use in Sodium Sulphur Batteries. US, US4456664.
- 5. Cava, S. and Tebcherani, S.M. (2007) Structural and Spectroscopic Characterization of Al2−xCrxO3 Powders Obtained by Polymeric Precursor Method. Journal of Sol-Gel Science and Technology, 43, 131-136. http://dx.doi.org/10.1007/s10971-007-1541-y
- 6. Nabi, H.S. and Pentcheva, R. (2011) Energetic Stability and Magnetic Coupling in (Cr1−xFex)2O3: Evidence for a Ferrimagnetic Ilmenite-Type Superlattice from First Principles. Physical Review B, 83, Article ID: 214424. http://dx.doi.org/10.1103/PhysRevB.83.214424
- 7. Yan, Y.F., Xu, X.Q., et al. (2013) Hot Corrosion Behaviour and Its Mechanism of a New Alumina-Forming Austenitic Stainless Steel in Molten Sodium Sulphate. Corrosion Science, 77, 202-209. http://dx.doi.org/10.1016/j.corsci.2013.08.003
- 8. Blacklocks, A.N., Atkinson, A., Packer, R.J., et al. (2006) An XAS Study of the Defect Structure of Ti-Doped α-Cr2O3. Solid State Ionics, 177, 2939-2944. http://dx.doi.org/10.1016/j.ssi.2006.08.028
- 9. Lavigne, O., Alemany-Dumont, C. and Normand, B. (2011) The Effect of Nitrogen on the Passivation Mechanisms and Electroni Properties of Chromium Oxide Layers. Corrosion Science, 53, 2087-2096. http://dx.doi.org/10.1016/j.corsci.2011.02.026
- 10. Shi, S.Q., Wysocki, A.L. and Belashchenko, K.D. (2009) Magnetism of Chromia from First-Principles Calculations. Physical Review B, 79, Article ID: 104404. http://dx.doi.org/10.1103/physrevb.79.104404
- 11. Corliss, L.M., Hastings, J.M., Nathans, R. and Shirane, G. (1965) Magnetic Structure of Cr2O3. Journal of Applied Physics, 36, 1099. http://dx.doi.org/10.1063/1.1714118
- 12. Saalfeld, V.H. (1964) Strukturuntersuchungen im System Al2O3-Cr2O3. Zeitschrift für Kristallographie, 120, 342-348. http://dx.doi.org/10.1524/zkri.1964.120.4-5.342
- 13. Qi, W.H., Wang, M.P. and Xu, G.Y. (2003) The Particle Size Dependence of Cohesive Energy of Metallic Nanoparticles. Chemical Physics Letters, 372, 632-634. http://dx.doi.org/10.1016/S0009-2614(03)00470-6
- 14. Wu, M.M., Jiang, Y., Wu, Y., et al. (2011) Structural Elastic and Electronic Properties of Mg(Cu1−xZnx)2 Alloys Calculated by First-Principles. Journal of Alloys and Compounds, 509, 2885-2890. http://dx.doi.org/10.1016/j.jallcom.2010.11.148
- 15. Zhou, D.W., Liu, J.S. and Xu, S.H. (2010) Thermal Stability and Elastic Properties of Mg3Sb2 and Mg3Bi2 Phases from First-Principles Calculations. Physica B, 405, 2863-2868. http://dx.doi.org/10.1016/j.physb.2010.04.013
*通讯作者。