Advances in Clinical Medicine
Vol.
10
No.
01
(
2020
), Article ID:
33836
,
7
pages
10.12677/ACM.2020.101001
Genotype Analysis of OPHN1 Gene Mutation in Non-Specific Mental Retardation Family with Literature Review
Huikun Duan, Li Wang, Shuang Hu, Zhouxian Bai, Xiangdong Kong*
Genetic and Prenatal Diagnosis Center, The First Affiliated Hospital of Zhengzhou University, Zhengzhou Henan
Received: Dec. 12th, 2019; accepted: Dec. 30th, 2019; published: Jan. 6th, 2020

ABSTRACT
Objective: To detect genetic causes of mental retardation in a Chinese mental retardation family in Henan Province. Method: A mental retardation pedigree was recruited and the proband was performed high-throughput sequencing to detect the intellectual impairment-related genes, and then the mutations in the candidate genes were verified by Sanger sequencing. Result: High-throughput sequencing results suggested that the proband carried two suspected pathogenic sites. They were OPHN1gene c.1973T>G(p.L658W) and GRIN2B gene c.190G>T(p.V64L), and Sanger sequencing was used to verify the two mutation sites in the proband. Sanger sequencing result also showed that the mother of the proband carried the heterozygous c.190G>T(p.V64L) in GRIN2B gene but had no symptoms. This did not meet the pathogenesis of the autosomal dominant disease, so the causing possibility of GRIN2B gene c.190G>T(p.V64L) site was ruled out. The mother and grandmother of the proband both carried the heterozygous variation c.1973T>G(p.L658W) in OPHN1 gene, and the sister and maternal aunt did not carry OPHN1 gene c.1973T>G(p.L658W) mutation. This was conformed to X-linked recessive inheritance. Conclusion: The mutation c.1973T>G(p.L658W) in OPHN1 gene was a novel mutation, which may be the cause of mental retardation in this non-specific mental retardation family. High-throughput sequencing combined with Sanger sequencing technology can diagnose mental retardation disease efficiently and accurately.
Keywords:Mental Retardation, Gene Mutation, High-Throughput Sequencing, OPHN1 Gene

一例非特异性精神发育迟滞家系OPHN1基因突变分析并文献复习
段会坤,王莉,胡爽,白周现,孔祥东*
郑州大学第一附属医院遗传与产前诊断中心,河南 郑州
收稿日期:2019年12月12日;录用日期:2019年12月30日;发布日期:2020年1月6日

摘 要
目的:对一个中国河南省精神发育迟滞家系进行致病基因突变筛查。方法:收集1个精神发育迟滞家系,应用高通量测序技术对先证者进行智力障碍相关基因检测,发现可疑致病基因后,采用Sanger测序技术对先证者亲属进行突变筛查,确定该家系的可疑致病位点。结果:高通量测序结果提示先证者携带两个可疑致病位点:OPHN1基因c.1973T>G(p.L658W)半合子变异和GRIN2B基因c.190G>T(p.V64L)杂合变异,并进行了一代测序验证。Sanger测序结果显示先证者母亲携带GRIN2B基因c.190G>T(p.V64L)杂合变异,由于母亲未发病,不符合常染色体显性遗传疾病发病机制,排除此位点致病的可能性。先证者母亲和外祖母均携带OPHN1基因c.1973T>G(p.L658W)杂合变异,先证者妹妹和姨妈未携带OPHN1基因c.1973T>G(p.L658W)变异,符合X-连锁隐性遗传方式。结论:OPHN1基因c.1973T>G(p.L658W)变异为新的变异,可能是该非特异性精神发育迟滞家系的致病原因,高通量测序结合Sanger测序方法可以高效准确地对该病进行基因诊断。
关键词 :精神发育迟滞,基因突变,高通量测序,OPHN1基因

Copyright © 2020 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 引言
精神发育迟滞(Mental retardation, MR)是一组由多种不同病因,包括遗传、感染、中毒、营养缺乏、早期环境刺激等引起的疾病,其主要特征为智力低下和社会适应困难,其中25%~50%的MR是由于遗传因素所致,包括染色体异常、基因拷贝数异常和单个基因突变 [1] [2]。由遗传因素引起的MR可分为特异性精神发育迟滞(Syndromic Mental Retardation, SMR)和非特异性精神发育迟滞(Non-specific Mental Retardation, NSMR),前者有明显的临床体征,往往与遗传综合征相伴随,而后者一般没有明显的临床体征,仅表现为认知功能障碍,无其他脑形态结构上的改变或别的临床特征 [3]。据文献报道世界MR的患病率为1%~3% [4] [5]。2000年我国抽样调查显示0~6岁儿童智力残疾现患率为0.931%,年均发现率为1.331%,每年新增13.6万例,其中2/3原因不明 [6]。近年来,随着人类基因组研究的快速发展,与精神发育迟滞相关的基因及致病位点相继被发现克隆,精神发育迟滞的分子遗传学分析已成为该病诊断的重要辅助手段。高通量测序技术可以一次性对多个基因进行测序,目前已经广泛应用于寻找疾病的候选基因上。本研究中我们对河南省1例精神发育迟滞先证者进行了高通量测序,并经过Sanger测序验证及携带者筛查,发现了该家系的可疑致病基因及其变异,先将结果报道如下。
2. 资料与方法
2.1. 研究对象
2019年3月来自河南安阳的1个精神发育迟滞核心家系(见图1),就诊于郑州大学第一附属医院遗传与产前诊断中心。先证者,男,4岁,表现为脑瘫,不会说话,不能行走,全身肌张力低,生活不能自理。先证者父母、妹妹、姨妈、外祖父母表型正常,先证者两个舅舅为精神发育迟滞患者,已经去世。经遗传咨询后,留取先证者外周血进行高通量测序,找到疑似致病基因后,留取先证者外祖母、母亲、姨妈、妹妹的外周血进行验证及携带者筛查。本研究经郑州大学第一附属医院医学伦理委员会批准,所有基因检测均经过家属的同意并签署知情同意书。
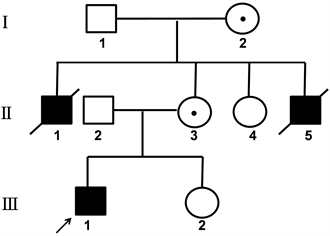
Figure 1. The family tree of non-specific mental retardation, the arrow shows the proband
图1. 1个非特异性精神发育迟滞家系图谱,箭头所示为先证者
2.2. 方法
2.2.1. DNA提取
签署知情同意后,EDTA-K2抗凝管采集先证者及其亲属外周血2 ml,用Omega DNA提取试剂盒提取基因组DNA,DNA总量5 μg以上,−20℃保存备用。
2.2.2. NGS测序和数据分析
利用目标区域捕获测序法对先证者264个已知与智力障碍相关的致病基因外显子及外显子-内含子交接区进行检测,获得高通量测序原始数据,之后去除低质量的数据,利用BWA (Burrows wheeler aligner)将“干净的”读序与人类基因组参考序列(UCSC, hgl9)比对。本实验中每个样品基因的平均测序深度约为118×,捕获区覆盖率达99%以上。为了找出致病性的点突变,进一步对所有检测基因的碱基变异进行筛选,在dbSNP147、Hapmap和千人基因组(1000 Genomes)等数据库上进行比对,排除已报道过的SNPs。针对错义变异应用polyphen_2、SIFT、MutationTaster等数据库对变异致病性进行生物信息学预测分析。新突变的命名参照国际基因变异命名体制(http://www.hgvs.org/mutnomen)提供的命名法命名。
2.2.3. PCR扩增 和Sanger测序验证
对先证者及其家属进行Sanger测序验证及携带者筛查,参考Ensembl数据库(http://www.ensembl.org)中OPHN1基因序列(ENST00000355520.5)和GRIN2B基因序列(ENST00000609686.3),应用GeneTool软件设计引物,由上海生工合成。OPHN1基因第21号外显子及其侧翼的引物序列,上游: 5'-TTTTGTGGGATATAGTTGGTTTGT-3',下游:5'-TCCACTTAAATATATCTCCAAACTCA-3'。GRIN2B基因第2号外显子及其侧翼的引物序列,上游:5'-GCTGGTAGATGGAGTTGGGT-3',下游: 5'-GGCTTCCTGGTCTGTGTCAT-3'。PCR反应体系成分为:2×反应缓冲液Mix(Mg2+ Plus) 13 μL,TaKaRa Taq酶1 μL,基因组DNA (50 ng/μL) 1 μL,引物(25 pmol/μL)各1 μL,补水至26 μL。扩增反应条件为95℃预变性4 min,95℃变性30 s,复性温度55℃~68℃退火30 s,72℃延伸30 s,共32个循环后,72℃延伸7 min。PCR产物用1.5%的琼脂糖凝胶电泳检测,确定产物大小与预期一致。之后PCR产物送上海生工进行Sanger双向测序,测序结果用Chromas软件进行比对分析,寻找基因突变位点。
3. 结果
3.1. NGS测序结果
对家系中先证者264个智力障碍疾病相关基因各外显子编码区域及剪接区的序列变异情况进行高通量测序,发现两个可疑致病位点:1) 在OPHN1基因第21外显子发现c.1973T>G (p.L658W)半合子变异,可导致X-连锁隐性疾病。该变异为错义突变,在HGMD专业版数据库中未见报道。经SIFT软件预测为有害的,经polyphen_2和MutationTaster预测为良性,GERP++值为5.05,比较保守。此变异在dbSNP147、Hapmap和千人基因组数据库未收录,在千人基因组数据库中出现的频率极低,不属于多态性位点。2)另外在GRIN2B基因第2外显子发现c.190G>T(p.V64L)杂合变异,可导致常染色体显性遗传疾病。该变异为错义突变,在HGMD专业版数据库中未见报道。经MutationTaster软件预测为有害的,经polyphen_2为良性,GERP++值为5.63,比较保守,此变异虽在dbSNP147中为rs150070901,但在千人基因组数据库未收录,在千人基因组数据库中出现的频率极低。
3.2. GRIN2B基因Sanger测序结果
对先证者GRIN2B基因变异进行Sanger测序验证,发现先证者携带GRIN2B基因第2外显子c.190G>T(p.V64L)杂合变异,与NGS测序检测结果一致(图2(A))。之后采用Sanger测序方法,分析先证者母亲GRIN2B基因c.190G>T(p.V64L)位点的变异情况,结果发现先证者母亲同样携带GRIN2B基因c.190G>T(p.V64L)杂合变异(图2(B))。由于GRIN2B基因变异导致的早发幼儿癫痫性脑病27型或智力低下6型均为常染色体显性遗传,而且先证者母亲自述无智力低下症状,不符合表型与基因型共分离,因此排除GRIN2B基因c.190G>T(p.V64L)变异为该家系致病原因的可能性。
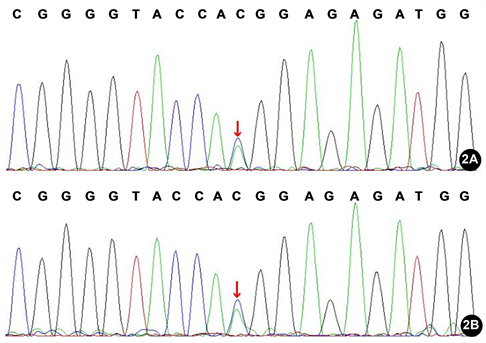 A:先证者,携带GRIN2B基因c.190G>T(p.V64L)杂合变异(箭头,反向测序);B:先证者母亲,携带GRIN2B基因c.190G>T(p.V64L)杂合变异(箭头,反向测序)。
A:先证者,携带GRIN2B基因c.190G>T(p.V64L)杂合变异(箭头,反向测序);B:先证者母亲,携带GRIN2B基因c.190G>T(p.V64L)杂合变异(箭头,反向测序)。
Figure 2. The sequencing results of gene mutation in 2exon of GRIN2B
图2. GRIN2B基因第2外显子Sanger测序图
3.3. OPHN1基因Sanger测序结果
对先证者进行Sanger测序验证,发现先证者为OPHN1基因第21外显子c.1973T>G(p.L658W)半合子变异,与NGS测序检测结果一致(图3(A))。该变异导致第658位亮氨酸变为色氨酸,为错义突变。该位点无相关致病性报道,为新突变,不是已知正常SNP位点,经SIFT软件预测为有害,推测c.1973T>G(p.L658W)变异可能为该先证者的致病原因。接下来采用Sanger测序方法对先证者亲属进行携带者筛查,分析家系中母亲OPHN1基因c.1973T>G(p.L658W)位点的变异情况,结果发现先证者母亲为OPHN1基因c.1973T>G(p.L658W)杂合变异携带者(图3(B))。由于OPHN1基因变异导致的X连锁非特异性精神发育迟滞为X-连锁隐性遗传,推测OPHN1基因c.1973T>G(p.L658W)变异为该精神发育迟滞家系致病变异的可能性大。另外对先证者外祖母、妹妹、姨妈进行了携带者筛查,发现先证者外祖母为OPHN1基因c.1973T>G(p.L658W)杂合变异携带者(图3(C)),再加上先证者两个舅舅均为智力低下患者(已故),再次证明了OPHN1基因c.1973T>G(p.L658W)变异为该家系的致病变异的可能性大。携带者筛查结果显示先证者姨妈和妹妹未携带OPHN1基因c.1973T>G(p.L658W)变异(图3(D)、图3(E))。
 A:先证者,携带OPHN1基因c.1973T>G(p.L658W)半合子变异(箭头,反向测序);B:先证者母亲,携带OPHN1基因c.1973T>G(p.L658W)杂合变异(箭头,反向测序);C:先证者外祖母,携带OPHN1基因c.1973T>G(p.L658W)杂合变异(箭头,反向测序);D:先证者妹妹,未携带OPHN1基因c.1973T>G(p.L658W)变异(箭头,反向测序);E:先证者姨妈,未携带OPHN1基因c.1973T>G(p.L658W)变异(箭头,反向测序)。
A:先证者,携带OPHN1基因c.1973T>G(p.L658W)半合子变异(箭头,反向测序);B:先证者母亲,携带OPHN1基因c.1973T>G(p.L658W)杂合变异(箭头,反向测序);C:先证者外祖母,携带OPHN1基因c.1973T>G(p.L658W)杂合变异(箭头,反向测序);D:先证者妹妹,未携带OPHN1基因c.1973T>G(p.L658W)变异(箭头,反向测序);E:先证者姨妈,未携带OPHN1基因c.1973T>G(p.L658W)变异(箭头,反向测序)。
Figure 3. The sequencing results of gene mutation in 21exon of OPHN1
图3. OPHN1基因第21外显子Sanger测序图
4. 讨论
在本研究中,我们对1个中国汉族X-连锁非特异性精神发育迟滞家系进行了分析,应用NGS对先证者进行智力障碍相关基因检测,在OPHN1基因编码区上发现一个新的变异c.1973T>G(p.L658W),该变异为该家系的可疑致病变异;然后,我们采用Sanger测序方法对先证者母亲和外祖母进行OPHN1基因变异来源检测,发现先证者母亲和外祖母均携带该位点杂合变异,符合X-连锁隐性遗传的疾病发病机制,推测该变异为先证者的致病原因,也可能为先证者两个舅舅(智力障碍,已故)的致病变异。
迄今为止,共有8个X连锁非特异性精神发育迟滞相关的基因被鉴定,包括FMR2、GDI1、OPHN1、PAK3、ARHGEF6、IL1RAPL、TM4SF2和FACL4 [7]。OPHN1基因是1997年法国科学家Bienvenu等在一位患有轻度精神发育迟滞并且具有X;12平衡易位的女性病人中发现的 [8]。1998年,Billuart及其同事通过定位克隆和RT-PCR 等方法将OPHN1基因定位在Xq12 [9]。OPHN1基因大小约390 kb,编码802个氨基酸的RhoGTP酶激活蛋白(RhoGAP),RhoGAP结构域可激活Rho、Rac1和Cdc42等3种GTP酶的活性,参与调节RhoGTP酶的信号传导过程。OPHN1蛋白包括两个结构域:RhoGTP酶激活结构域和氨基末端结构域,而OPHN1蛋白与F-肌动蛋白相互作用的最小结构域位于蛋白质的C末端 [10]。OPHN1基因内的错义突变、小的缺失及插入都会影响信号传导过程,使大脑处理信息的能力被限制,从而导致精神发育迟滞。目前发现OPHN1基因变异可导致X-连锁精神发育迟滞综合征 [11],小脑发育不全 [12],发育迟缓 [13] 等疾病。本研究中,先证者临床表现为脑瘫,4岁不会说话,不能行走,全身肌张力低,生活不能自理等,该患儿临床表现与文献报道相符。
目前,在人类基因突变数据库(HGMD)已发现OPHN1基因突变34种。大片段缺失重复突变(部分或整个基因)约占47%,点突变和微小缺失或插入突变占53%,其中错义突变和无义突变大约占26%。OPHN1基因第21外显子c.1973T>G(p.L658W)变异可以导致第658位亮氨酸变为色氨酸,为错义突变,该位点无相关致病性报道,为新的突变。此变异在dbSNP147和千人基因组数据库均未收录,在千人基因组数据库中出现的频率极低,不是已知正常SNP位点,经SIFT软件预测为有害,推测c.1973T>G(p.L658W)变异可能为该先证者的致病原因。对此变异进行了来源验证,发现先证者c.1973T>G(p.L658W)变异来自母亲,母亲的变异遗传自外祖母,而且先证者两个舅舅(智力障碍,已故)很可能也遗传了c.1973T>G(p.L658W)变异而发病,符合基因型与表型共分离原则,符合X-连锁隐性遗传病的特点,为该精神发育迟滞家系致病变异的可能性大。但是,此位点为新的变异,本研究未对其进行功能研究,其致病性还需进一步验证。
精神发育迟滞家族史是儿童精神发育迟滞的最大风险因素,即家庭成员中有精神发育迟滞的患者,其后代患病的风险大大增加。因此应在结婚或孕前进行遗传咨询与指导,同时有遗传风险的家系需要进行产前诊断,而明确病因是产前诊断的关键。高通量测序技术结合Sanger测序方法可以高效准确地对精神发育迟滞患者进行基因诊断,有助于尽早发现精神发育迟滞患儿,从而预防出生缺陷。
基金项目
郑州大学第一附属医院院内青年基金。
文章引用
段会坤,王 莉,胡 爽,白周现,孔祥东. 一例非特异性精神发育迟滞家系OPHN1基因突变分析并文献复习
Genotype Analysis of OPHN1 Gene Mutation in Non-Specific Mental Retardation Family with Literature Review[J]. 临床医学进展, 2020, 10(01): 1-7. https://doi.org/10.12677/ACM.2020.101001
参考文献
- 1. Coe, B.P., Witherspoon, K., Rosenfeld, J.A., et al. (2014) Refining Analyses of Copy Number Variation Identifies Specific Genes Associated with Developmental Delay. Nature Genetics, 46, 1063-1071.
https://doi.org/10.1038/ng.3092 - 2. Pariltay, E., Durmaz, A., Durmaz, B., et al. (2014) Genome-Wide Copy Number Variation Analysis in Idiopathic Intellectual Disability/Multiple Congenital Anomalies. Genetic Counseling, 25, 221-229.
- 3. Gecz, J. and Mulley, J. (2000) Genes for Cognitive Function: Developments on the X. Genome Research, 10, 157-163.
https://doi.org/10.1101/gr.10.2.157 - 4. Leonard, H. and Wen, X. (2002) The Epidemiology of Mental Retardation: Challenges and Opportunities in the New Millennium. Mental Retardation and Developmental Disabilities Research Reviews, 8, 117-134.
https://doi.org/10.1002/mrdd.10031 - 5. Shaffer, L.G. (2005) American College of Medical Genetics Guideline on the Cytogenetic Evaluation of the Individual with Developmental Delay or Mental Retardation. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 7, 650-654.
https://doi.org/10.1097/01.gim.0000186545.83160.1e - 6. 王新宪, 朱庆生. 中国0-6岁残疾儿童抽样调查报告[M]. 北京: 中国统计出版社, 2001: 94-103.
- 7. Nawara, M., Bal, J. and Mazurczak, T. (2002) Monogenic Causes of Nonspecific X-Linked Mental Retardation Molecular Aspects. Medycyna Wieku Rozwojowego, 6, 281-294.
- 8. Bienvenu, T., Der-Sarkissian, H., Billuart, P., et al. (1997) Mapping of the X-Breakpoint Involved in a Balanced X;12 Translocation in a Female with Mild Mental Retardation. European Journal of Human Genetics: EJHG, 5, 105-109.
https://doi.org/10.1159/000484743 - 9. Billuart, P., Bienvenu, T., Ronce, N., et al. (1998) Oligophrenin-1 Encodes a rhoGAP Protein Involved in X-Linked Mental Retardation. Nature, 392, 923-926.
https://doi.org/10.1038/31940 - 10. Fauchereau, F., Herbrand, U., Chafey, P., et al. (2003) The RhoGAP Activity of OPHN1, a New F-Actin-Binding Protein, Is Negatively Controlled by Its Amino-Terminal Domain. Molecular and Cellular Neurosciences, 23, 574-586.
https://doi.org/10.1016/S1044-7431(03)00078-2 - 11. Zanni, G., Saillour, Y., Nagara, M., et al. (2005) Oligophrenin 1 Mutations Frequently Cause X-Linked Mental Retardation with Cerebellar Hypoplasia. Neurology, 5, 1364-1369.
https://doi.org/10.1212/01.wnl.0000182813.94713.ee - 12. Philip, N., Chabrol, B., Lossi, A.M., et al. (2003) Mutations in the Oligophrenin-1 Gene (OPHN1) Cause X Linked Congenital Cerebellar Hypoplasia. Journal of Medical Genetics, 40, 441-446.
https://doi.org/10.1136/jmg.40.6.441 - 13. Classen, C.F., Riehmer, V., Landwehr, C., et al. (2013) Dissecting the Genotype in Syndromic Intellectual Disability Using Whole Exome Sequencing in Addition to Genome-Wide Copy Number Analysis. Human Genetics, 132, 825-841.
https://doi.org/10.1007/s00439-013-1296-1
NOTES
*通讯作者。