Hans Journal of Computational Biology
Vol.06 No.03(2016), Article ID:18411,9
pages
10.12677/HJCB.2016.63006
Research on Statistical Analysis of Gene Splicing Sites
Hongbin Li*, Guangzhong He
Medical School, Xianyang Vocational and Technical College, Xianyang Shaanxi

Received: Aug. 5th, 2016; accepted: Aug. 19th, 2016; published: Aug. 26th, 2016
Copyright © 2016 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



ABSTRACT
The genes of eukaryotes are composed of several exons and introns. After transcript process, sequences of exons are retained, while sequences of introns are cleaved off. A large number of experiments of molecular biology validate that the splicing sites between exon and intron follow the rule of GT-AG, only a few GT or AG sequences are true splicing sites, and the accuracy of the prediction still needs to be improved. In this study, the training dataset of splicing site of HS3D was downloaded, and a statistical analysis of the sequence near the splicing site of the promoter was carried out. The sequence showed high specificity when the true and false sequence lengths of the left splicing site side and right splicing site side were both more than seven, which was helpful to train the sequences characters so as to accurately identify the true and false splicing sites.
Keywords:Gene, Splice Site, Statistical Analysis

基因剪切位点的统计分析研究
李宏彬*,赫光中
咸阳职业技术学院医学院,陕西 咸阳

收稿日期:2016年8月5日;录用日期:2016年8月19日;发布日期:2016年8月26日

摘 要
真核生物的基因由若干外显子和内含子交替组成,外显子序列在转录后保留,而内含子序列转录过程中被剪切掉。大量分子生物学实验验证基因的剪切位点遵从GT-AG规则,然而只有很少的含GT或AG序列是真剪切位点,目前预测的准确程度仍有待提高。本研究下载了HS3D剪切位点训练数据集,对启动子剪切位点附近的序列进行了统计分析研究。当真、假序列长度在剪切位点左旁和右旁均超出各七个位点时,序列呈现很高的特异性,可以使用这些特异性序列作为特征进行训练,从而准确地识别真假剪切位点。
关键词 :基因,剪切位点,统计分析

1. 引言
基因组学是研究基因序列结构、功能分析和如何利用基因的一门学科,而基因剪切位点识别是其中重要的研究方向之一。真核生物基因是由一段段编码区和非编码区碱基序列嵌合而成,编码区又称为外显子(exon),它们之间的非编码区被称为内含子(Intron),基因的首尾还各有一段具备一定功能的非编码区,分别称为启动子和终止子。外显子和内含子的大小变化不定,内含子一般要远长于外显子。真核基因先转录为前mRNA (包含所有外显子和内含子序列),然后序列中的内含子需要被除去而外显子相互链接为成熟的mRNA,这个过程称为剪接(Splicing),如图1所示。成熟的mRNA每个三联体核苷酸构成一个密码子,将被翻译成一个氨基酸,它们决定了蛋白质的氨基酸线性顺序。因此,如果剪切不够准确,如多出或缺少一个核苷酸,下游经翻译的密码子就会出误,最终生成错误的蛋白质。大量实验数据表明绝大多数剪切位点遵从GT-AG规则(极少数个例显示遵从AT-AC规则),外显子-内含子连接区呈现高度保守性,也就是在内含子序列的5’端(从外显子过渡到内含子)特征为GT,而在其3’端(从内含子过渡到外显子)特征为AG,然而海量基因组测序数据显示满足GT-AG规则的序列绝大多数并不是真内含子序列。基因剪接位点常用的研究方法有:人工神经网络 [1] 、隐马尔可夫模型 [2] 、动态规划 [3] 、支持向量机 [4] 、贝叶斯网络 [5] 和频谱3-周期性 [6] 等。
2. 方法和分析
HS3D (Homo Sapiens Splice Sites Dataset) [7] 是意大利Pollastro从GeneBank DNA序列数据库中提取的基因剪接位点序列数据集,数据集中的每个条目记录剪切位点从上游到下游总长为140个字符的DNA序列数据,剪接符均遵从GT-AG规则,GT位于位点71到72,AG位于位点69到70。数据集分为四个部分:真EI (exon to intron)、假EI、真IE (intron to exon)和假IE,真EI和真IE记录的数据条目数相对较少,分别为2796和2880个,而假EI和假IE数据条目数相对极多,分别为271,928和329,360个。为了观察真假剪切位点临近序列的差异,本研究依据HS3D数据集中的数据,对140个位点真、假EI和IE (的四种碱基(A、T、C、G)出现频率进行了比较,分别如图2、图3所示。其中横坐标代表临近序列位置,纵坐标表示某位置的真(左)、假(右) EI、IE碱基A (红)、T (绿)、C (蓝)或G (黄)出现频率(0到1之间)。从图2、图3中可以观察到,总体来说,从剪接点上游位点到下游位点,真剪接位点临近序列的碱基呈现随位置变化的出现频率,特别在剪接特征符左右十个位置(统计数据见表1),而假的除特征符GT和AG左右一两个位置以外,在其余位点呈现近似接近的碱基出现频率。真EI序列在剪接位点的一致序列
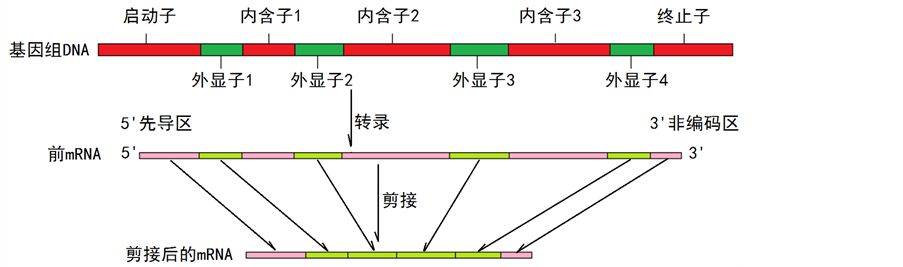
Figure 1. Structure, transcription and splicing of eukaryotic gene
图1. 真核基因的结构、转录和剪接

Table 1. The base frequency comparison in sites from −10 to 10 between true splicing sequences and false splicing sequences
表1. 真、假剪切位点序列−10到10各碱基出现频率比较
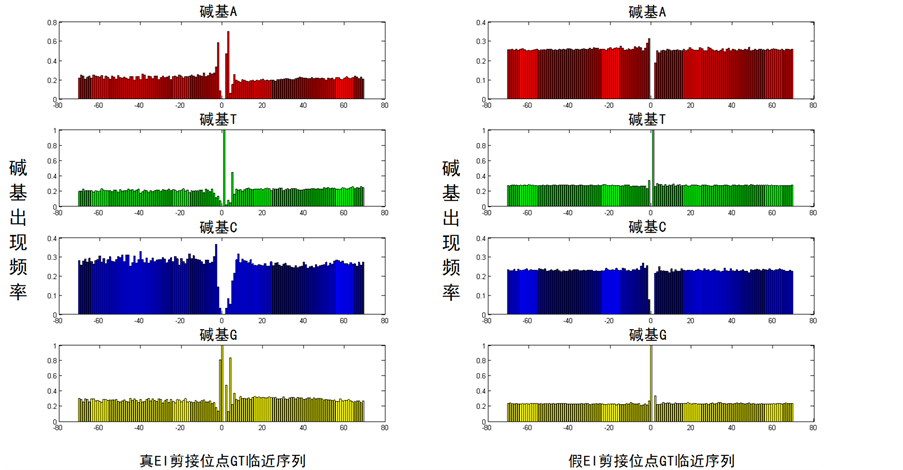
Figure 2. The base statistical frequency comparison of sequence close to splicing site between true EI dataset and false EI dataset
图2. 真、假EI剪接位点临近序列碱基统计频率比较

Figure 3. The base appearance frequency comparison of sequence close to splicing site between true IE dataset and false IE dataset
图3. 真、假IE剪接位点临近序列碱基出现频率比较
是MAG GT RAG (M: A/C, R: A/G)真IE序列在剪接位点的一致序列是YYNC AG RN (Y: T/C, N: A/T/C/G)。
参考真EI、IE数据集,本研究对以剪切位特征符(GT、AG)为中心的碱基六聚体出现百分频率进行了统计,若总频率为1,则真EI和真IE频度靠前的六聚体EI、IE数据集中真剪接位点多聚体出现频率数据分别如表2和表3所示,这些六联体总频度占真EI和真IE数据集中样本量的绝大多数,分别为82.53%
Table 2. The top 24 frequency statistic of 6-mer base close to splicing sites in true EI dataset
表2. 真EI剪接位点六聚体碱基出现频率统计(前24个)
Table 3. The top 63 frequency statistic of 6-mer close to splicing sites in true IE dataset
表3. 真IE剪接位点六聚体出现频率统计(前63个)
和82.65%。统计发现,剪切位特征符左右各有两个碱基核苷酸,应有4的4次方(256)中可能性,数据集中真,EI和IE剪接六聚体至少出现一次分别占118和181种可能性,而假EI和IE剪接六聚体覆盖所有的256种可能性,反映剪接位点临近序列的特异性。本研究依次对EI、IE数据集中真、假剪接位点多聚体重合度统计,包括6聚体、8聚体(4096种可能性,特征符前三后三)、10聚体(65,536种可能性,特征符前四后四)、12聚体(1,048,576种可能性,特征符前五后五)和14聚体(16,777,216种可能性,特征符前六后六),如图4所示。图中反映6聚体、8聚体和12聚体,真假剪接聚体的重合度很高(重合是指,某个位于剪接位点的多聚体,若真、假数据集中都至少出现1例,则该聚体重合),若依此聚体序列为特征进行识别会导致极高的错误率,而14聚体以上真、假聚体序列的重合度大幅度下降,特异性显著增强,有利于以此为特征进行训练和进行真剪接位点识别判断。
那么总长相同的聚体,不同的选取方式,会对重叠率有什么影响呢?本研究做了一个实验,这些多聚体总长相同,但在剪接特征符GT或AG前后选取的核苷酸数目不同,然后统计真、假多聚体的重叠率,结果如表4所示,其中示例EI和IE数据集中真、假剪接位点前五后五模式多聚体分布比较分别如图5和图6所示。十二聚体中,选取方式前6后4 (前6 GT后4)在EI真、假数据集中获得最低的重叠率,前2后8 (前2 AG后8)在IE真、假数据集中获得最低的重叠率。十四聚体中,选取方式前8后4 (前8 GT后4)在EI真、假数据集中获得最低的重叠率,前3后9 (前3 AG后9)在IE真、假数据集中获得最低的重叠率。获得最低重叠率的十二聚体位置与十四聚体接近,但十二聚体重叠率远高于十四聚体,如果以十二聚体作为特征进行真、假剪接位点识别,虽然特征碱基数目少、速度快,但会产生过高的错误率,因此真假剪接位点识别训练应选择十四聚体以上作为特征模式。
Table 4. The overlap rate comparison between true and false dataset of 12-mer and 14-mer close to splicing sites by different selection methods
表4. 剪接临近十二和十四聚体在不同选取方式下真、假数据集的重叠率比较

Figure 4. The N-mer overlap rate statistic of true and false splicing site in EI and IE dataset
图4. EI、IE数据集中真、假剪接位点多聚体重合度统计

Figure 5. The N-mer (five before and five after GT site) distribution comparison between true and false splicing site in EI dataset
图5. EI数据集中真、假剪接位点GT前五后五模式多聚体分布比较

Figure 6. The N-mer (five before and five after AG site) distribution comparison between true and false splicing site in IE dataset
图6. IE数据集中真、假剪接位点AG前五后五模式多聚体分布比较
3. 结论
本文使用公共数据库HS3D的序列数据对基因剪接位点的序列进行了统计分析。通过统计获得真假剪接位点的碱基出现频率,分析反映真剪接位点临近序列的碱基呈现随位置变化的出现频率,而假的除特征符GT和AG左右一两个位置以外,在其余位点呈现近似接近的碱基出现频率。通过统计还获得了占数据库中绝大多数的真剪接位点EI和IE六联体序列。研究还发现14聚体以上,真剪切位点临近序列的特异性显著增强,这有利于以此序列为特征进行训练,从而准确地识别真假剪切位点。
致谢
感谢陕西省科技厅社会发展科技攻关项目基金(2016SF-343)资助。
文章引用
李宏彬,赫光中. 基因剪切位点的统计分析研究
Research on Statistical Analysis of Gene Splicing Sites[J]. 计算生物学, 2016, 06(03): 41-49. http://dx.doi.org/10.12677/HJCB.2016.63006
参考文献 (References)
- 1. Sun, J. (1993) Predicting the Splicing Sites of mRNA by Neural Network. Acta Biophysica Sinica, 9, 127-131.
- 2. Xia, H., Zhou, Q. and Yanda, L.I. (2002) Application of Hidden Markov Model in the Recognition of Splicing Sites. Journal of Tsinghua University, 42, 1214-1217.
- 3. Snyder, E.E. and Stormo, G.D. (1993) Identification of Coding Regions in Genomic DNA Sequences: An Application of Dynamic Programming and Neural Networks. Nucleic Acids Research, 21, 607-613. http://dx.doi.org/10.1093/nar/21.3.607
- 4. Zhang, L.R. and Luo, L.F. (2003) Splice Site Prediction with Quadratic Discriminant Analysis Using Diversity Measure. Nucleic Acids Research, 31, 6214-6220. http://dx.doi.org/10.1093/nar/gkg805
- 5. Cai, D., Delcher, A., Kao, B. and Kasif, S. (2000) Modeling Splice Sites with Bayes Networks. Bioinformatics, 16, 152-158. http://dx.doi.org/10.1093/bioinformatics/16.2.152
- 6. Yin, C. and Yau, S.T. (2007) Prediction of Protein Coding Regions by the 3-Base Periodicity Analysis of a DNA Sequence. Journal of Theoretical Biology, 247, 687-694. http://dx.doi.org/10.1016/j.jtbi.2007.03.038
- 7. Pollastro, P. and Rampone, S. (2002) HS3D, a Dataset of Homo Sapiens Splice Regions, and Its Extraction Procedure from a Major Public Database. International Journal of Modern Physics C, 13, 1105-1117. http://dx.doi.org/10.1142/S0129183102003796
*通讯作者。