Hans Journal of Computational Biology
Vol.07 No.02(2017), Article ID:21068,10
pages
10.12677/HJCB.2017.72002
A Predictor of Protein Secondary Structure Based on a Continuously Updated Templet Library
Pengjie Zhou1, Ming Wen2, Peisheng Cong1*, Tonghua Li1*
1School of Chemical Science and Engineering, Tongji University, Shanghai
2College of Chemistry and Chemical Engineering, Central South University, Changsha Hunan
*通讯作者。

Received: May 30th, 2017; accepted: Jun. 19th, 2017; published: Jun. 22nd, 2017

ABSTRACT
Protein secondary structure prediction is an important field of computational biology. Although the accuracies of the existed state-of-the-art approaches are more than 80% but these methods have a common limitation. They couldn’t learn new structure knowledge of currently measured proteins, and couldn’t change the used model and their parameters. Thus, they couldn’t satisfy our expecting in the changing world. Here, we present a predictor of protein secondary structure based on a continuously updated templet library: SIPSS. The basic stone of our approach is structural similarity based on sequence homology. First, a continuously updated templet library is constructed, which can automatically download the measured protein structure data from PDB per-month. After screening, the new information of protein sequences and structures are supplied into the template library. Then a query sequence is aligned against the template library by using PSI BLAST, and a new variable-SPSSM variable is obtained. Last, the SPSSM variable is used in a conditional random field algorithm for modelling and prediction. Our experiments showed that SIPSS can online learn new protein structure information and its prediction accuracy (80.6%) of protein secondary structure measured in recent times is significantly better than the state-of-the art approaches. SIPSS is available free of charge at http://cheminfo.tongji.edu.cn/SIPSS/.
Keywords:Protein Secondary Structure, Prediction, Continuously updated, SPSSM Variable, Conditional Random Field
基于可持续更新模板库的蛋白质二级 结构预测器
周鹏杰1,文明2,丛培盛1*,李通化1*
1同济大学化学科学与工程学院,上海
2中南大学化学化工学院,湖南 长沙

收稿日期:2017年5月30日;录用日期:2017年6月19日;发布日期:2017年6月22日

摘 要
蛋白质二级结构预测是计算生物学研究的重要领域。虽然现有优秀的机器学习方法的预测准确度已经超过80%,但是它们都有共同的缺陷:不能及时学习最新实测的蛋白质结构信息,不能持续修改模型和参数,从而满足人们在日新月异时代对蛋白质二级结构预测的要求。本文构建了基于可持续更新模板库的蛋白质二级结构预测器:SIPSS。我们的新方法以同源序列的结构保守性为基本原理。首先我们建立了一个可持续更新的模板库,每月自动从蛋白质数据库中下载新测定的蛋白质结构数据,经过筛选将新的序列和结构信息补充进模板库。然后对于查询序列,用多重同源比对与模板库比对,得到新的变量:SPSSM变量。最后,我们以SPSSM为变量,用条件随机场建模和预测。实际测试表明,SIPSS能够在线学习新的蛋白质结构信息,对新近测定的蛋白质二级结构预测准确度(80.6%)明显高于现有的预测器。SIPSS网站:http://cheminfo.tongji.edu.cn/SIPSS/,可供用户免费使用。
关键词 :蛋白质二级结构,预测,持续更新,SPSSM变量,条件随机场

Copyright © 2017 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 引言
蛋白质二级结构预测一直是计算生物学研究的热点领域 [1] 。蛋白质二级结构预测往往是蛋白质立体结构和功能预测的第一步,高质量的蛋白质二级结构预测不但为随后研究提供坚实的基础,而且很大程度上影响其后研究结果的准确度。
近年来,蛋白质二级结构预测的主流方法是机器学习方法。机器学习的预测准确的也从最初的50%左右,提高到超过80%,一些优秀的蛋白质二级结构预测网络服务器,如PSIPRED [2] 、Porter [3] 、Jpred [4] 、SPIDER [5] 和DeepCNF [6] 等,预测准确度已经接近Rost预言的预测理论极限88% [7] 。分析这些前人的研究成果,我们可以看到两条清晰的技术发展思路:一条是改进原有方法,或者运用新的计算方法。比如人工神经网络 [8] 、支持向量机 [9] 、隐马尔科夫 [10] 、条件随机场 [11] 、深度学习 [12] 等等。新的机器学习方法往往能有效提高预测的准确度。另一条研究思路是提出新的变量。比如各种序列信息、氨基酸物理化学新性质,溶剂可及性、位置特异性得分矩阵(Position-Specific Scoring Matrix, PSSM) [13] 等。PSSM是蛋白质序列多重同源比对的结果,通常用PSI-BLAST [14] 获得,对于每一个氨基酸,对应着20个元素的矢量。PSSM反映了序列进化信息,也反映了氨基酸保守和突变的信息。PSSM的一个直观结果是同样一种氨基酸如果出现在一条序列不同的位置,它们的PSSM可能是不同的。这就意味着氨基酸的PSSM反映了此氨基酸所处位置的周围环境。PSSM的这个特点,使得它成为蛋白质二级结构预测的最重要变量。
我们组在前期的研究中提出一种蛋白质二级结构预测的新变量,结构位置特异性得分矩阵(Structural Position-Specific Scoring Matrix, SPSSM) [15] 。SPSSM与PSSM类似,是通过多重序列比对得到的,不过它是三个元素的矢量,对应于二级结构的三种状态:螺旋,折叠和无规卷曲。首先,我们构造了一个采自NCBI非冗余900多万条序列的与BLAST兼容的模板库。这个模板库的条目存储两类信息,一类是序列信息,即蛋白质的氨基酸组成;一类是对应的二级结构信息。对应的结构信息是通过PSI-BLAST比对蛋白质结构数据库PDB (Protein Data Bank) [16] 得到的。然后,在训练建模和预测的时候,查询序列用PSI-BLAST比对模板库,取出N条匹配序列以及对应的结构信息,计算SPSSM。最后,我们用PSSM和SPSSM共23个变量构筑了新型蛋白质二级结构预测器,SPSSMPred,取得了非常优秀的预测准确度。随后,我们又将这种策略扩展到8态二级结构预测,构建了SPSSM8 [17] ,同样显著提高了预测准确度。
不可否认,SPSSMPred和SPSSM8是有缺陷的:当预测序列找不到同源模板或同源序列片段时,SPSSM变量的三个元素是零,我们的预测器只能靠PSSM变量了。其必然结果是预测准确度很低。事实上所有机器学习方法都有这种缺陷。设想人们怎么可能准确预测与建模序列完全无关序列的结构呢?国际蛋白质预测中心CASP (Critical Assessment of Techniques for Protein Structure Prediction)竞赛的组织者将蛋白质结构预测方法分为基于模板(Template-Based Method, TBM)和非模板(Template-Free Method, FM)两类方法 [18] 。一般认为,TBM计算快,准确度较高,但外延不足;FM计算费用大且准确度较低,但对于完全陌生的序列可能提供有用结构信息。
我们的时代是大数据时代,新测定的蛋白质结构数据不断涌现。过去无法解析的蛋白质,如膜蛋白,线粒体蛋白等的结构随着实验仪器的更新、实验技术的丰富也不断被破译。由于有些新测定的蛋白与PDB库存储的蛋白同源性较低,现有的蛋白质二级结构预测器大都不能准确预测它们的二级结构,更无法及时利用它们的结构信息去预测其它类似蛋白。现行的解决的办法是只能是经过一段时间(往往3~5年),重新收集数据,重新建模,发布预测器新的版本。面对数据的滚滚洪流,现行的办法显然是笨拙的、低效的。
本文提出基于可持续更新模板库的蛋白质二级结构预测新方法:SIPSS (Sustainable Inferring Protein Secondary Structure)。SIPSS以同源序列结构保守性为理论基础,用结构位置特异性得分矩阵作为变量,用条件随机场算法进行建模和预测。结构位置特异性得分变量是基于可持续更新的模板库获得。它能够随着PDB发布的最新蛋白质实验结构数据自动更新和补充新的模板。这样SIPSS就有能力及时学习实验获得的最新成果,所以能够准确地预测那些有序列同源性蛋白的3态二级结构。
2. 材料和方法
2.1. 数据
数据准备:蛋白质序列从PDB下载。其中DB-1为2015年前发布的蛋白质序列,序列相似度 < 90%,共41350条序列;DB-2为2015年1月01日到2016年3月31之间公布的、序列相似性 < 90%的序列,共5061条序列。
模板库:初始模板库由DB-1获得。经过PISCES [19] 去冗余(设置条件:序列相似度 < 90%、序列长度超过40个氨基酸、分辨率 < 3.0 Å,和R值 < 0.3),得到21553条序列。模板库是可BLAST数据库。模板库的规模会随着时间不断增大。模板库中的序列的理论二级结构用DSSP [20] 程序计算,少数序列中计算不出二级结构的个别氨基酸删除(比如无序氨基酸)。DSSP给出的8态二级结构转化为3态二级结构,即α螺旋(H)、β折叠(E)和无规卷曲(C)。
训练集:将DB-1和DB-2合并,经PISCES去冗余(设置条件:序列相似度 < 25%、序列长度超过40个氨基酸、分辨率 < 3.0 Å,和R值 < 0.3),得到9676条序列。其中来源于DB-1作为训练集,共8507条序列。
测试集:上述去冗余结果中来源于DB-2的作为测试集(1169条序列),这样处理,保证训练集与测试集的序列相似性小于25%。
CASP12:CASP的测试序列是对所有从事蛋白质结构预测的挑战,它们与PDB的冗余度很小(<30%),主要用于三级结构预测竞赛。我们下载了成文时PDB发布的条目,经自我去冗余90%后,得到了20条非冗余序列作为CASP12。训练集,测试集和CASP12序列的二级结构也由DSSP计算得到。
2.2. 结构位置特异性得分矩阵
结构位置特异性得分矩阵是我们先前提出的非常有效的蛋白质二级结构预测的变量,它只有3个元素。对于查询序列来讲,首先用PSI-BLAST与模板库进行同源比对。然后选取N条(默认值为10)匹配序列所对应的二级结构。最后根据匹配序列的二级结构,按照查询序列氨基酸计算第i个SPSSM变量的第s个元素:
 (1)
(1)
式中,j是匹配的序列数。P(i,j,s)是第j条匹配序列第i个氨基酸、结构为s的布尔值,即等于s为1,其它为0。对单个氨基酸而言,SPSSM变量是归一化的。
SPSSM变量充分利用了同源序列的结构相似性,从已知结构的实验数据中获得二级结构的可能性信息。它个数少,运算快,效果明显,是十分优秀的结构预测变量。SPSSM变量已被证实能够显著提高二级结构预测的准确度。所以本文的二级结构预测器只采用SPSSM变量。
2.3. 可持续更新的模板库
为了及时处理大数据时代源源不断涌现的数据流,在机器学习领域,人们提出了在线学习(Online Learning)的策略 [21] 。在线学习方法认为,人们可以不需要重新训练预测模型,只要利用新数据,部分修改模型中变量的权重,就可以修正模型,达到提高准确度的目的。我们经过反复演算,提出了更简单的办法:更新模板库。我们发现,只要持续更新模板库,二级结构预测的准确度就能保持高水平,而重新建模,大约只能再提高0.1%左右。这很大程度上是由SPSSM变量所致。因此采用可持续更新的模板库可以在不降低预测准确度的前提下大大简化预测器的复杂性。
可持续更新的模板库的设计如下:在初始模板库基础上,根据PDB发布新蛋白结构实验数据的频率,下载当月所有蛋白质序列及其结构文件。经过PISECES去冗余后(设置条件:序列相似度 < 90%、序列长度 < 40个氨基酸、分辨率 < 3.0 Å,和R值 < 0.3)。它们的二级结构由DSSP计算。然后用当前的模板库计算它们的SPSSM变量,并预测其二级结构。如果一条序列的预测准确度高于我们预先设定的阈值(默认值为88%),说明当前的模板库含有与这条序列高度相似的条目,完全能代表这条序列。因此该序列被丢弃。反之,如果一条序列的预测准确度小于阈值,则该序列及其结构信息补充进模板库。毫无疑问,这些新蛋白信息进入模板库后,预测这些蛋白序列及其形似序列的准确度会大大提高。图1是更新模板库的运行框图。
2.4. SIPSS蛋白质二级结构预测器
我们的蛋白质二级结构预测器名为SIPSS。无论建模还是预测,首先,查询序列与模板库进行同源比对,计算并记录最佳匹配得分(Score)以及SPSSM变量。其次,依据SPSSM变量用条件随机场(Conditional Random Field, CRF) [22] 建模或者预测。然后,输出预测结果并根据Score对预测结果给出参考评价,即预测系列与模板库的同源性,分高(high,score大于360),中(medium)低(low,score小于60)三种,用户可据此判断预测结果的质量。
预测器由Web网站和后端预测程序组成,网站由用java+JSP编写,使用SQL Server储存数据,负责接受用户提交的蛋白序列;后端用C#语言编写。模板库更新程序用Python和C#语言编写。模板库每月更新一次,保证SIPSS能够使用最新的蛋白质结构信息为用户服务。SIPSS输入是FASTA格式的待测序列,输出是可自行下载二级结构预测结果。
2.5. 评价指标
二级结构预测的准确度有许多评价指标,其中最常用的是Qs和SOVs (Segment Overlap Score) [23] 。SOV度量预测与实验二级结构片段的覆盖程度,被认为是比单个氨基酸准确度更重要的指标。Qs定义是:
 (2)
(2)
式中,s表示二级结构类型,即H,E,C或者3 (所有类型)。

Figure 1. The flowsheet of the continuously updated templet library
图1. 持续更新模板库流程图
SOVs定义为:
 (3)
(3)
式中,N是所有s类型氨基酸数。minov和maxov分别表示括号内最小和最大值,sobs和spred分别表示s类型连续片段实验和预测氨基酸的个数。Len()表示长度。 取4个数的最小值,即
取4个数的最小值,即
 (4)
(4)
3. 结果与讨论
3.1. 同源序列的结构保守性
SIPSS预测能力主要依靠SPSSM变量,而SPSSM变量是同源比对模板库的结果。因此我们预测方法的理论基础是同源序列的结构保守性。实际上,所有机器学习方法都建立在同源序列的结构保守性之上。一方面,我们无法要求机器学习方法能够准确预测一条完全与训练集不同序列的二级结构;另一方面,我们可以将所有蛋白质序列看做是20种氨基酸组成的“同源”序列。关键问题是同源片段的长短,同源片段短,比如小于3个氨基酸片段,对应的二级结构保守性很差,根本无法用于预测。采用长的同源片段,比如氨基酸数目大于9,结构保守性是可以保证了,但匹配的训练样本却找不到了,因此也无法用于预测。多重同源比对(PSI-BLAST)用动态规划很好地解决了这个问题 [14] 。它采用打分(e value等),氨基酸取代(BLOSUM-62)和多次循环等,保证了查询同源序列的质量。我们的实验证实,PSI-BLAT找到的同源序列片段,二级结构是高度保守的,可以提高二级结构预测的准确度 [15] 。
我们用训练集(8507条序列)建模,初始模板库(21553条序列)做比对库,预测测试集(1169条序列)。由于测试集与训练集及初始模板库的序列同源性很低(<25%),整体预测准确度Q3约为72%。仔细分析预测的各条序列,我们看到如果一条待预测序列能在初始模板库找到同源序列或同源序列片段,预测准确度一般都能超过90%。如果一条待预测序列找不到同源序列片段,预测准确度就降低到50%~60%左右,预测水平与现有的优秀预测器差不多(参见表2)。
测试集的预测准确度改善的办法是增加变量,比如溶剂可及性,伪氨基酸编码等,但是增加改进效果不明显且额外增加计算费用。而用更新模板库却效果明显。一旦我们用2016年3月更新模板库,训练模型不变,预测准确度Q3超过94%。因此SIPSS最终采用SPSSM变量和能够持续更新的模板库。
3.2. 自动更新模板库的预测结果
我们以上述训练模型和2016年3月模板库为基础,测试SIPSS的性能。每月程序自动下载PDB发布的新蛋白质序列及其结构。根据设定的条件自动更新模板库。从2016年4月到12月的更新前后预测结果见表1。
从表中我们可以看到,每月大约有1百条左右的新序列与先前模板库冗余度较低。这些序列及其结构加入模板库后预测准确度明显提高。
SIPSS是一个简洁的预测器,它不能准确预测与模板库序列同源性很低蛋白的二级结构,但是它能及时学习新的蛋白质结构知识,是新颖的有在线学习能力的蛋白质二级结构预测器。
3.3. 与现有预测器的比较
由于采用可持续更新的模板库,SIPSS有很强的自学能力,能够随着时间推移,不断学习新的知识,增强预测能力。我们用上述训练模型,初始模板库、2016年8月的模板库和12月模板库,对CASP12

Table 1. Comparison of performances between before and after updating the template library
表1. SIPSS更新模板库前后预测结果比较
注:年月后面的括号内数据是当月预测准确度低于0.88的蛋白条数。
的测试目标进行预测,准确度Q3分别为62.9% (SOV3:54.9),79.1% (73.4)和94.4% (92.4)。为了进行比较,我们选了5个优秀的且被广泛使用的二级结构预测器:DeepCNF [6] ,SPIDER2 [5] ,PSIPRED3 [2] ,Jpred4 [4] 以及Porter4 [3] 。这些预测器在发布的当时是十分优秀的,并且随后又不断改进,发布了新版本。PSIPRED是第一个用PSSM预测二级结构的预测器 [24] ,它现在可用版本是3.3。Jpred和Porter都有了新的4版本。SPIDER2采用交叉预测的方法提高了预测的准确度。DeepCNF则用深度学习的方法以序列和PSSM为变量,显著改进了预测准确度。上述5种方法以及我们用2016年8月模板库预测结果列于表2。
从预测结果我们可以看出,如果是用初始模板库,我们的方法是不好的。但是随着时间推移,我们的更新模板库逐渐包含了CASP12的预测序列或者相似序列。SIPSS的预测准确度不断提高,到2016年8月就超越其它所有预测器。这说明任何机器学习的预测器如果不及时改变模板和模型,很快将失去它的优势。比如DeepDNF对CASP10和CASP11的Q3都超过84%,但对于CASP12只有72.9%。而我们的SIPSS具有在线学习能力,能够及时跟上新的变化,能准确地推演出我们已知几乎所有蛋白质的二级结构。
3.4. 新蛋白的预测例子
SIPSS预测器不但能够准确预测已学习的序列二级结构,它还能利用序列同源性,预测新蛋白的二级结构。我们用2016年12月的模板库预测2017年1月25日发布的整合素αvβ6:5FFG [25] 。整合素是细胞表面跨膜受体家族,在信号传递,调控和许多生命活动中起重要作用。5FFGb蛋白链有257氨基酸,SIPSS预测Q3是88.4%,预测结果如图2表示。
Table 2. Comparison of different predictors on CASP12
表2. 不同预测器对CASP12预测结果比较
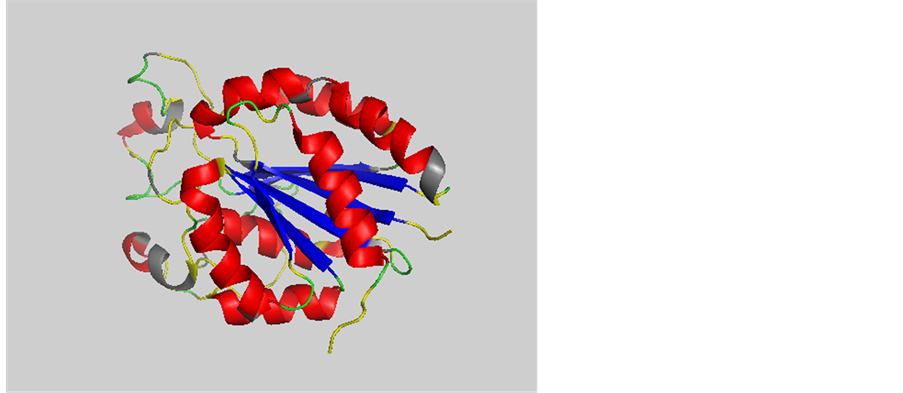
Figure 2. The diagram of the predicted protein 5FFGb structure. Where, red shows the correct predicted α helixes; green shows the correct predicted coils; blue shows the correct predicted β sheets; grey shows the incorrect predicted structures; yellow shows the disorder structures
图2. 蛋白质5FFGb预测结果示意图。图中,红色代表已正确预测的α螺旋类型。绿色代表已正确预测的无规卷曲类型。蓝色代表正确预测的β折叠类型。灰色表示错误预测的结构。黄色代表从DSSP是计算不出的二级结构。5ffgb三维结构是用Pymol画的
4. 结论
本文构建了基于可持续更新模板库的蛋白质二级结构预测器。与前人不同之处是我们从同源序列结构保守性出发,仅用3个元素的SPSSM变量以及可持续更新的模板库来预测蛋白质二级结构。我们的SIPSS预测器不需要频繁更改版本就能随时间自动更新,利用最新的结构知识去预测蛋白质的二级结构。实验表明SIPSS能够在任何时候准确的预测人们已经知道的绝大多数蛋白的二级结构,我们相信它在计算机预测蛋白质结构领域将会有广阔的应用前景。
下一步的工作我们将研究自动更新模型的方法,完善SIPSS,以达到更高的蛋白质二级结构预测准确度。本文的工作可以看作是人工智能–增强学习(reinforcement learning)的初步尝试。我们相信随着人工智能技术在计算生物学中不断探索和应用,人们一定能够构建完美的蛋白质结构预测器。
基金项目
国家自然科学基金资助项目(21275108)。
文章引用
周鹏杰,文明,丛培盛,李通化. 基于可持续更新模板库的蛋白质二级结构预测器
A Predictor of Protein Secondary Structure Based on a Continuously Updated Templet Library[J]. 计算生物学, 2017, 07(02): 13-22. http://dx.doi.org/10.12677/HJCB.2017.72002
参考文献 (References)
- 1. Rost, B. (2001) Review: Protein Secondary Structure Prediction Continues to Rise. Journal of Structural Biology, 134, 204-218. https://doi.org/10.1006/jsbi.2001.4336
- 2. Buchan, D.W.A., et al. (2013) Scalable Web Services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Research, 41, 349-357. https://doi.org/10.1093/nar/gkt381
- 3. Mirabello, C. and Pollastri, G. (2013) Porter, Pale Ale 4.0: High-Accuracy Prediction of Protein Secondary Structure and Relative Solvent Accessibility. Bioinformat-ics, 29, 2056-2058. https://doi.org/10.1093/bioinformatics/btt344
- 4. Drozdetskiy, A., et al. (2015) JPred4: A Protein Second-ary Structure Prediction Server. Nucleic Acids Research, 43, 389-394. https://doi.org/10.1093/nar/gkv332
- 5. Heffernan, R., et al. (2015) Improving Prediction of Secondary Structure, Local Backbone Angles, and Solvent Accessible Surface Area of Proteins by Iterative Deep Learning. Scientific Reports, 5, Article ID: 11476. https://doi.org/10.1038/srep11476
- 6. Wang, S., et al. (2016) Protein Secondary Structure Prediction Using Deep Convolutional Neural Fields. Scientific Reports, 6, 355-378. https://doi.org/10.1038/srep18962
- 7. Rost, B. and Sander, C. (1993) Prediction of Protein Secondary Structure at Better than 70% Accuracy. Journal of Molecular Biology, 232, 584-599. https://doi.org/10.1006/jmbi.1993.1413
- 8. Cai, Y.D., et al. (2002) Artificial Neural Network Method for Predicting Protein Second-ary Structure Content. Computers & Chemistry, 26, 347-350. https://doi.org/10.1016/S0097-8485(01)00125-5
- 9. Kieslich, C.A., et al. (2016) CONSSERT: Consensus SVM Model for Accurate Prediction of Ordered Secondary Structure. Journal of Chemi-cal Information & Modeling, 56, 455-461. https://doi.org/10.1021/acs.jcim.5b00566
- 10. Kim, H. and Park, H. (2003) Protein Secondary Structure Prediction Based on an Improved Support Vector Machines Approach. Protein Engineering, 16, 553-560. https://doi.org/10.1093/protein/gzg072
- 11. Liu, Y., et al. (2004) Comparison of Probabilistic Combination Methods for Protein Sec-ondary Structure Prediction. Bioinformatics, 20, 3099-3107. https://doi.org/10.1093/bioinformatics/bth370
- 12. Spencer, M., Eickholt, J. and Cheng, J. (2014) A Deep Learning Network Approach to ab Initio Protein Secondary Structure Prediction. IEEE/ACM Transactions on Computational Biology & Bioinformatics, 103-112.
- 13. Gribskov, M., Mclachlan, A.D. and Eisenberg, D. (1987) Profile Analysis: Detection of Distantly Related Proteins. Proceedings of the National Academy of Sciences of the United States of America, 84, 4355-4358. https://doi.org/10.1073/pnas.84.13.4355
- 14. Altschul, S.F., et al. (1997) Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Research, 25, 3389-3402. https://doi.org/10.1093/nar/25.17.3389
- 15. Li, D., et al. (2012) A Novel Structural Position-Specific Scoring Matrix for the Prediction of Protein Secondary Structures. Bioinformatics, 28, 32-39. https://doi.org/10.1093/bioinformatics/btr611
- 16. Rose, P.W., et al. (2015) The RCSB Protein Data Bank: Views of Structural Biology for Basic and Applied Research and Education. Nucleic Acids Research, 43, 345-356. https://doi.org/10.1093/nar/gku1214
- 17. Cong, P., et al. (2013) SPSSM8: An Accurate Approach for Predicting Eight-State Second-ary Structures of Proteins. Biochimie, 95, 2460-2464.
- 18. Moult, J., et al. (2016) Critical Assessment of Methods of Protein Structure Prediction: Progress and New Directions in Round XI. Proteins-Structure Function & Bioinformatics, 84, 4-14. https://doi.org/10.1002/prot.25064
- 19. Wang, G. (2005) PISCES: Recent Improvements to a PDB Sequence Culling Server. Nucleic Acids Research, 33, W94-W98. https://doi.org/10.1093/nar/gki402
- 20. Kabsch, W. and Sander, C. (1983) Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen- Bonded and Geometrical Features. Biopolymers, 22, 2577-2637. https://doi.org/10.1002/bip.360221211
- 21. Hoi, S.C.H., Wang, J. and Zhao, P. (2014) LIBOL: A Library for Online Learning Algo-rithms. Journal of Machine Learning Research, 15, 495-499.
- 22. Lafferty, J.D., Mccallum, A. and Pereira, F.C.N. (2002) Conditional Random Fields: Probabilistic Models for Segmenting and Labeling Sequence Data. Proceedings of the Eighteenth International Con-ference on Machine Learning, 3, 282-289.
- 23. Zemla, A., Fidelis, K. and Rost, B. (1999) A Modified Definition of Sov, a Seg-ment-Based Measure for Protein Secondary Structure Prediction Assessment. Proteins-Structure Function & Bioinformatics, 34, 220-223. https://doi.org/10.1002/(SICI)1097-0134(19990201)34:2<220::AID-PROT7>3.0.CO;2-K
- 24. Jones, D.T. (1999) Protein Sec-ondary Structure Prediction Based on Position-Specific Scoring Matrices. Journal of Molecular Biology, 292, 195-202. https://doi.org/10.1006/jmbi.1999.3091
- 25. Dong, X., et al. (2017) Force Interacts with Macromolecular Structure in Activation of TGF-β. Nature, 542, 55-59. https://doi.org/10.1038/nature2103