International Journal of Psychiatry and Neurology
Vol.05 No.04(2016), Article ID:18968,4
pages
10.12677/IJPN.2016.54010
A Case Report of Sporadic Creutzfeldt-Jakob Disease
Baoliang Wang, Hongbo Pang, Wei Jin, Shizao Fei*
Department of Neurology, The Affiliated No. 2 People’s Hospital of Wuhu, Wannan Medical College, Wuhu Anhui
Received: Oct. 27th, 2016; accepted: Nov. 15th, 2016; published: Nov. 18th, 2016
Copyright © 2016 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



ABSTRACT
Creutzfeldt-Jakob disease (CJD) is one of the fatal human prion diseases, which results in dementia as well. Most of CJD are sporadic. CJD often meets misdiagnosis because of its atypical symptom especially in its early stage. Here we report a case of sporadic CJD with hypomnesis as initial symptom.
Keywords:Creutzfeldt-Jakob Disease, Diagnosis, 14-3-3 Protein, Imaging

散发型克雅病1例报告
王保良,庞洪波,金炜,费世早*
皖南医学院附属芜湖市第二人民医院神经内科,安徽 芜湖

收稿日期:2016年10月27日;录用日期:2016年11月15日;发布日期:2016年11月18日

摘 要
克雅病是人类朊蛋白病的一种,属致死性疾病,也是进展性痴呆的常见病因之一,多为散发型;由于其早期症状不典型,易导致误诊。本文报道一例以记忆力减退为首发症状的散发型克雅病。
关键词 :克雅病,诊断,14-3-3蛋白,影像学

1. 引言
克雅病(Creutzfeldt-Jakob disease,CJD),又称皮质-纹状体-脊髓变性,是人类朊蛋白病的一种,本病既有遗传性又有传染性,是进展性痴呆的常见病因之一。全球发病率大约是百万分之一,国内于1980年首次报道 [1] 。CJD分为散发型(sCJD)、医源型(获得型)、遗传型和变异型四种类型,其中sCJD约占90%,本病早期诊断较困难,容易误诊和漏诊。本文现报道1例临床诊断为sCJD的病例。
2. 病例分析
患者,男性,60岁,因“记忆力减退6个月,行走不稳10余天”入院。患者自2015年7月份开始于无明显诱因下出现记忆障碍,出门后常找不到家,同时有脾气暴躁、多疑、言语减少等性格改变,后在我市精神病院就诊,具体治疗不详,病情逐渐加重,出现视力减退、阵发性左上肢不自主抖动、不能交流等症状,近10天出现行走不稳,无法直线行走,伴有小便失禁。起病后去多家医院就诊,均未明确诊断。于2016年1月7日至我院就诊。病程中无发热、头痛,无恶心、呕吐,既往无高血压、糖尿病史,无家族遗传病史。入院查体:神志恍惚,精神萎靡,记忆力、定向力、理解力减退,语言功能检查不配合,双侧瞳孔等大等圆,直径约2.5 mm,直接、间接对光反射灵敏,双侧眼球活动自如,无眼震,双侧鼻唇沟对称,伸舌、示齿不配合,深浅感觉、共济运动检查不配合,四肢能动,肌力无法测,肌张力正常,深浅反射正常,颈软、无抵抗,脑膜刺激征阴性,双侧病理征阴性,尿便失禁。人院后积极完善相关检查:血、尿、便常规,生化,肝、肾功能,凝血功能,CRP,胸部CT,心电图等均未见异常;脑脊液压力、常规、生化、脑脊液找霉菌、脑脊液找抗酸杆菌、脑脊液培养未见异常;脑电图提示异常脑电图(患者检查时躁动不安,不能配合);头颅MRI + DWI示双侧顶枕叶、额叶、及双侧丘脑异常信号(图1)。入院后拟诊“中枢神经系统感染,朊蛋白病?”予以抗病毒、支持、对症等处理,病情仍进行性加重,入院第15天患者开始陷入昏迷,频繁出现四肢抖动,肌张力显著增高,脑脊液送检国家疾病预防控制中心检测提示l4-3-3蛋白阳性。
3. 结果
住院20天后患者家属签字自动出院,随访得知,患者于出院后7天死亡。根据以上症状体征及辅助检查结果,临床诊断为sCJD。
4. 讨论
CJD是由朊蛋白感染所致的一种致命性的中枢神经系统变性疾病,发病的主要原因是由于正常细胞的PrPC转变成了引起人类及动物朊蛋白病的PrPSC,PrPSC沉积在脑组织中,从而导致脑的海绵状变性和胶质细胞增生 [2] 。不同类型CJD临床表现有所不同,但大部分CJD患者都有高级神经功能减退的表现,其中sCJD好发于50~70岁人群,两性患病率相当,常有大脑皮层、锥体系、锥体外系、小脑受损的症状与体征,sCJD的诊断常采用以下标准 [3] ,1) 确定诊断:脑组织活检检测到PrPSC者;2) 疑似诊断:进行性痴呆、典型的脑电图改变,和(或)脑脊液l4-3-3蛋白阳性,且病程短于2年,以及符合下列4种中的至少2种:①肌阵挛;②视力障碍;③小脑症状;④无动性缄默;3) 可能诊断:仅有进行性痴呆,且病程短于2年,以及符合下列4种中的至少2种:①肌阵挛;②视力障碍;③小脑症状;④无动性缄默。

 (a) (b)
(a) (b)
 (c) (d)
(c) (d)
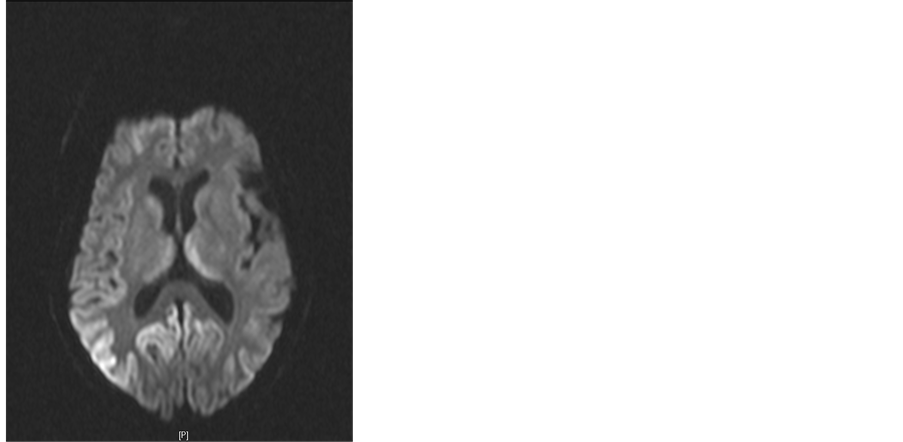
Figure 1. Patients with brain MRI showed no abnormal T1WI (a), FLAIR and DWI showed high signals of bilateral parietal occipital lobe, frontal lobe and bilateral thalamus, mainly in the right hemisphere ((b), (c)), Enhancement no abnormality found (d)
图1. 患者头颅MRI检查T1WI未见异常(a),FLAIR、DWI可见双侧顶枕叶、额叶及双侧丘脑异常高信号,以右侧半球为主((b), (c)),增强未发现异常(d)
遗憾的是,CJD的确诊有赖于脑组织活检。最新诊断标准强调了影像学检查在诊断CJD方面的重要价值。有证据表明 [4] ,磁共振序列中的DWI像以及FLAIR像在诊断CJD方面比脑电图更有价值,DWI和FLAIR诊断CJD的敏感性和特异性分别为91%~92.3%和93.8%~95%。近年来,对于sCJD的生物学标志物的检测方面取得了一定的进展,未来对于sCJD的诊断可能有所裨益 [5] 。
该例患者起病初期仅表现为记忆力减退,之后迅速发展为语言、行为、意识等高级精神功能障碍,并伴有锥体系、锥体外系及小脑损害表现,病程6个月时即不能交流,并很快陷入昏迷。结合脑脊液14-3-3蛋白阳性、异常脑电图以及MRI+DWI影像学特征,且该患者无CJD家族史,无感染源接触史及暴露史,排除其他痴呆相关疾病后,根据上述诊断标准,临床诊断为sCJD。
sCJD目前死亡率100%,临床上尚无有效的预防及治疗方法 [6] 。以朊蛋白形成的显性无效抑制作用为理论基础,正在进行基于此结构的药物研究,并且已经有了一些具有潜力的研究成果,特别是免疫治疗方面,已开始运用于动物实验及临床实验,但对其临床疗效尚需进一步进行验证 [7] [8] 。对于临床出现隐匿起病、缓慢进展的认知功能障碍,伴椎体系、椎体外系、小脑病变的患者,需考虑到CJD的可能,并及时予头颅MRI检查及脑脊液14-3-3蛋白检测。
文章引用
王保良,庞洪波,金 炜,费世早. 散发型克雅病1例报告
A Case Report of Sporadic Creutzfeldt-Jakob Disease[J]. 国际神经精神科学杂志, 2016, 05(04): 59-62. http://dx.doi.org/10.12677/IJPN.2016.54010
参考文献 (References)
- 1. 李梦东, 王宇明. 实用传染病学[M]. 第3版. 北京: 人民卫生出版社, 2004: 614-630.
- 2. Sikorska, B., Knight, R., Irenside, J.W., et al. (2012) Creutzfeldt-Jakob Disease. Advances in Experimental Medicine and Biology, 724, 76-90. http://dx.doi.org/10.1007/978-1-4614-0653-2_6
- 3. 王珍燕, 卢洪洲. 克雅病诊治[J]. 中国感染与化疗杂志, 2013, 13(5): 400-404.
- 4. 李泽, 李蒙燕, 郑浩. 散发型Creutzfeldt-Jakob病的临床、脑电图及头颅磁共振DWI影像特点研究[J]. 现代电生理学杂志, 2013, 20(3): 131-137.
- 5. Soomro, S. and Mohan, C. (2016) Biomarkers for Sporadic Creutzfeldt-Jakob Disease. Annals of Clinical and Translational Neurology, 3, 465-472. http://dx.doi.org/10.1002/acn3.304
- 6. Puoti, G., Bizzi, A., Forloni, G., et al. (2012) Sporadic Human Prion Diseases: Molecular Insights and Diagnosis. The Lancet Neurology, 11, 618-628. http://dx.doi.org/10.1016/S1474-4422(12)70063-7
- 7. Roettger, Y., Du, Y., Bacher, M., et al. (2013) Immunotherapy in Priori Disease. Nature Reviews Neurology, 9, 98-105. http://dx.doi.org/10.1038/nrneurol.2012.258
- 8. Burchell, J.T. and Panegyres, P.K. (2016) Prion Diseases: Immunotargets and Therapy. ImmunoTargets and Therapy, 5, 57-68. http://dx.doi.org/10.2147/ITT.S64795