Hans Journal of Surgery
Vol.04 No.01(2015), Article ID:14668,7
pages
10.12677/HJS.2015.41001
Identification Key Genes of Hepatocellular Carcinoma Base on TCGA Database
Junjun Jia, Ning He, Jing Zhang, Li Jiang, Yanfei Zhou, Lin Zhou, Shusen Zheng
Key Laboratory of Combined Multi-Organ Transplantation, Ministry of Public Health, Department of Hepatobiliary and Pancreatic Surgery, First Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou
Email: jiajunjun1987@163.com
Received: Nov. 3rd, 2014; revised: Nov. 20th, 2014; accepted: Dec. 5th, 2014
Copyright © 2015 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



ABSTRACT
Objective: Hepatocellular carcinoma (HCC) is a common cancer of the digestive system, is the third cause of death worldwide and the second cause of death in China. The Cancer Genome Atlas (TCGA) aims to better understand the molecular mechanisms of cancer by using a large-scale genome sequencing-based analysis techniques and extensive cooperation. This study introduces TCGA database to find key genes of HCC events. Materials and Methods: The data from TCGA were processed, integrated according to the standard procedure of TCGA, data types and levels were carefully assessed. Bioinformatics analysis was done using the DESeq and edgeR package of R language (3.1.1 version). Results were showed as pheatmap, VennDiagram, hist, PlotMA etc. Differences were defined as follows: expression increased more than two folds; P <0.05; gene ranked in the top 10%. Results: 17 mRNA chips of HCC and 9 mRNA chips of normal tissue were collected from TCGA data- base. Hist figure reflected the number of different gene was large. PLotMA map showed the distribution of gene expression, suggesting most genes of different expression were increased. 719 differentially expressed genes were found by DESeq, while 4413 by edger, among which 713 were common different genes. Conclusion: Compared to conventional microarray, TCGA method has its own advantages such as larger number of samples, less cost and easier for analyzing, offering opportunity for large-scale genomic studies of HCC and subsequent functional genomics-based research.
Keywords:Hepatocellular Carcinoma, TCGA, Chip
基于TCGA数据库的肝癌发生关键基因筛选
贾俊君,何宁,张静,姜骊,周燕飞,周琳,郑树森
浙江大学医学院附属第一医院肝胆胰外科,卫生部多器官联合移植研究重点实验室,杭州
Email: jiajunjun1987@163.com
收稿日期:2014年11月3日;修回日期:2014年11月20日;录用日期:2014年12月5日

摘 要
目的:肝癌是消化系统常见恶性肿瘤,是全世界第三位死亡原因和中国第二位死亡原因。肿瘤基因组图谱(TCGA)计划利用大规模测序为主的基因组分析技术,通过广泛的合作,理解癌症的分子机制,本研究利用TCGA数据库深入挖掘肝癌发生关键基因。材料方法:根据标准流程对TCGA数据进行处理、整合,对数据类型及水平进行评估,用R语言(3.1.1版本)中自带的DESeq和edgeR程序包进行分析,结果以热图(pheatmap)、韦恩图(VennDiagram)、hist、PlotMA等表示。差异基因的判断标准:1,表达量在2倍以上或者0.5倍以下,2,P < 0.05,3,基因排名在前10%。结果:TCGA数据库现有癌组织mRNA芯片信息17张,匹配正常组织mRNA芯片信息9张,共26张。Hist图反映的是每个经统计后P值得分布规律,图中可刊出P值接近0处频率很高,反映差异基因的数量很大。PLotMA图反应基因表达量的分布规律,提示表达上升基因数量较多。用DESeq方法一共找到719个差异基因,而用edgeR方法找到4413个差异基因,两种方法都鉴别出的共同差异基因713个。结论:TCGA法相较于传统的芯片筛选具有样本数量大、费用小、分析简单等优势,为更多的人进行大规模的肝癌基因组学研究以及基于基因组学的后续功能研究提供了可能性。
关键词 :肝癌,TCGA,芯片

1. 引言
肝癌是消化系统常见恶性肿瘤,是全世界第三位死亡原因 [1] 和中国第二位死亡原因 [2] 。现有的治疗手段不能有效预防与控制肝癌切除或者肝移植后复发,肝癌患者预后仍然不容乐观。因此需要进一步对肝癌发生及复发机制进行深入研究,寻找肝癌发生的关键基因及肝癌预后评估的生物学指标,以进一步改善患者预后。
肿瘤基因组图谱 (TCGA)计划由美国National Cancer Institute (NCI)和National Human Genome Re- search Institute (NHGRI)于2006年联合启动的项目,第一阶段为期三年,耗资1亿美元,研究的癌症类型包括多形性成胶质细胞瘤(GBM)、卵巢癌,并于2008年在Nature发表了GBM的研究成果,2009年9月,再投$2.75亿,针对20余种癌症进行大规模实验,目前总计36种癌症类型。TCGA利用大规模测序为主的基因组分析技术,通过广泛的合作,理解癌症的分子机制。提高人们对癌症发病分子基础的科学认识及提高我们诊断、治疗和预防癌症的能力。最终完成一套完整的与所有癌症基因组改变相关的“图谱”。本文着重介绍TCGA数据库及利用TCGA数据库现有的数据深入挖掘寻找肝癌发生的关键基因。
2. 材料与方法
2.1. TCGA数据处理流程
2.1.1. 组织处理
1) 癌症病人自愿捐赠肿瘤组织及正常组织样本,由人类癌症生物标本核心资源库承担癌症组织标本和正常组织标本的采集、处理和分配工作。
2) 组织样本经过严格标准处理(处理标准根据不同后续分析类型而异,具体标准参见 http://cancergenome.nih.gov/cancergenomics/tissuesamples),确保质量可以用于进一步分析及测序,并由相关中心(基因组测序中心 genome sequencing centers和肿瘤基因组鉴定中心cancer genome characterization centers)采用高通量测序技术进行基因和基因组排序。
3) 获得的临床资料中,可以识别病人身份的信息去掉。
2.1.2. 整合研究
1) TCGA基因组分析中心(GCC)比对肿瘤和正常组织,寻找异常的基因重组现象。
2) 高通量测序中心(GSC)分析与各癌症或者亚型相关的基因突变、扩增或者缺失。
3) 资料分析中心(GDAC)进行资料的整理、汇总、并提供图表报告给全体研究团队。
2.1.3. 资料分享
1) 资料综合中心(DCC)集中处理各个团队产生的资料,定期公开于网络上供全世界研究人员利用。
2) 提供公开的资料下载网站入口以方便进行资料搜索和下载。
2.2. TCGA数据类型和数据水平
2.3. TCGA数据分析方法
TCGA标准方法下载肝细胞肝癌癌症组织及正常组织信息,统计分析采用R语言(3.1.1版本)软件,需安装及加载的程序包(pheatmap,venndiagram,hist等),然后用DESeq和edgeR程序包进行分析,结果以热图(pheatmap)、韦恩图(VennDiagram)hist、PlotMA等表示。具体的差异基因分析策略参考oshlack等报道的方法[3] 。差异基因的判断标准:1——表达量在2倍以上或者0.5倍以下,2——P < 0.05,3——基因排名在前10%。
3. 结果
3.1. 数据检索
进入TCGA主页(http://cancergenome.nih.gov/)—Lunch Data Portal—Download Data—Data Matrix— Filter setting: select a disease (LIHC-liver hepatocellular carcinoma), Data Type(RNA Seq), platform: genome wide mRNA levels (Illumina mRNA-seq), microRNA levels (Illumina microRNA-seq), Tumor/Normal (tumor-matched or normal-matched) —Apply—Color cells by (tumor/nomal)—下载。
本次下载共得到癌组织芯片信息17张,正常组织芯片信息9张,共26张。
3.2. 表达谱差异基因
3.2.1. 基因分布
对所下载的26张芯片进行hist、plotMA分析结果见图1。Hist图反映的是每个统计后P值得分布规律,图中可看出P值接近0处频率很高,反映差异基因的数量很大。PLotMA图反应的是基因表达量的分布规律,图中红线代表与正常组织比较表达量无差异的基因,红线以上表示表达量升高的基因,反之表示表达量下降,由图可以看出大部分差异表达基因属于高表达。
3.2.2. 差异基因热图
分别用DESeq和edgeR程序包对下载的26张芯片信息进行热图(pheatmap)分析,结果见图2。由于

Table 1. Data types of TCGA
表1. TCGA数据类型
Table 2. Data levels and types of TCGA
表2. TCGA数据水平及类型
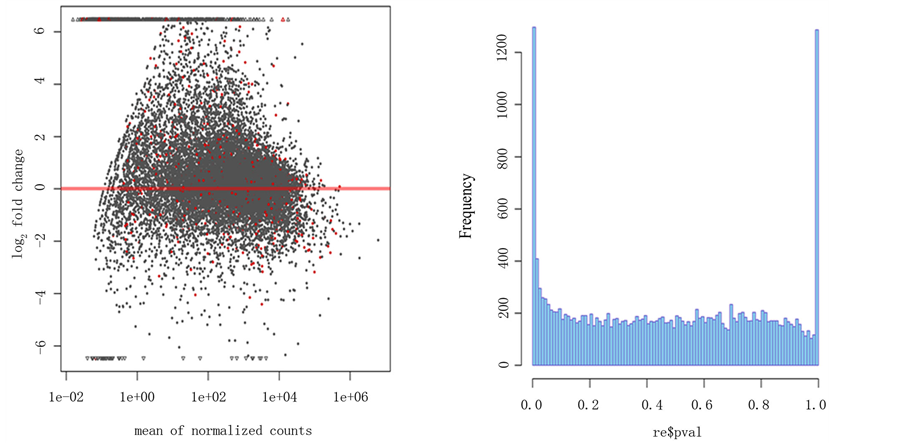 注:左图显示的PLotMA图,图中红线代表与正常组织比较表达量相同的基因,红线以上表示表达量升高的基因,反之表示表达量下降。
注:左图显示的PLotMA图,图中红线代表与正常组织比较表达量相同的基因,红线以上表示表达量升高的基因,反之表示表达量下降。
Figure 1. Diagrams of PloMA and hist
图1. PlotMA 和hist图
符合差异基因判断的基因较多,热图中右侧基因名称无法清晰显示,图3列出DESeq方法差异基因中的
 注:左图显示用DESeq方法找到的差异基因热图,右图显示用edgeR方法找到的差异基因热图。红色代表基因表达上调,绿色代表基因表达下调。
注:左图显示用DESeq方法找到的差异基因热图,右图显示用edgeR方法找到的差异基因热图。红色代表基因表达上调,绿色代表基因表达下调。
Figure 2. The heatmaps of differential genes by DESeq and edgeR
图2. 用DESeq和edgeR找到的差异基因热图
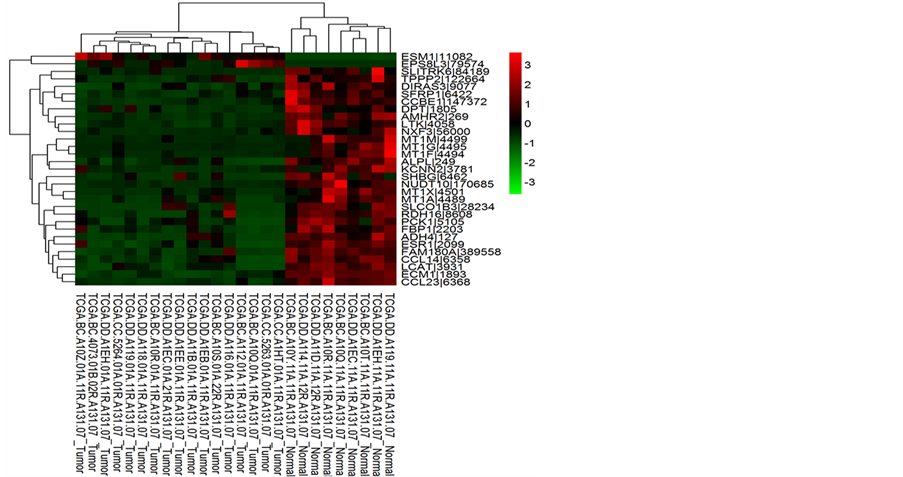 注:DEseq方法找到的差异基因中的30个基因热图。红色代表基因表达上调,绿色代表基因表达下调。
注:DEseq方法找到的差异基因中的30个基因热图。红色代表基因表达上调,绿色代表基因表达下调。
Figure 3. Thirty examples of differential genes
图3. 30个差异基因代表
30个。
3.2.3. 共同差异基因
图4显示的是用DESeq和edgeR方法寻找差异基因的韦恩图。图中我们可以看出用DESeq方法一共找到719个差异基因,而用edgeR方法找到4413个差异基因,两种方法都鉴别出的共同差异基因713个,包含三个表达下降(MT1B、BMP10和SYT10)和710个升高的基因(ALB、HP、FGB等)。
4. 讨论
我国是原发性肝细胞癌高发国家,占每年全球新发肝癌例数55%左右,死亡人数占全球的40%以上,而且肝癌的发生率在世界范围内呈现增加趋势[4] 。尽管以手术切除为核心的肝癌综合治疗体系已经形成,但是术后肿瘤高复发率仍是威胁患者长期生存的主要因素[5] [6] 。此外,对于肿瘤晚期,无法实施手术的患者目前也尚无理想的治疗方案。近年来,随着对肿瘤发生机制的深入认识,以此为基础开发的分子靶向药物在临床逐渐应用,这给晚期肝癌患者带来了希望的同时也存在药物疗效欠佳和副作用大的问题。因此,探索肝细胞癌新的发病机制、寻找新的治疗靶点有极其重要的临床和科学意义。
美国政府发起的癌症和肿瘤基因图谱(TCGA)计划,试图通过应用基因组分析技术,特别是采用大规模的基因组测序,将人类全部癌症的基因组变异图谱绘制出来,并进行系统分析,旨在找到所有致癌和抑癌基因的微小变异,探索癌细胞发生、发展的机制,进一步研发新的诊断和治疗方法,最后勾画出整个新型“预防癌症的策略”。TCGA以人类基因组计划(HGP)为基础,研究癌症过程中基因组的改变。但两者关注的焦点不同,HGP专注于疾病的遗传因素(先天因素),TCGA更关心人类出生后细胞中的基因
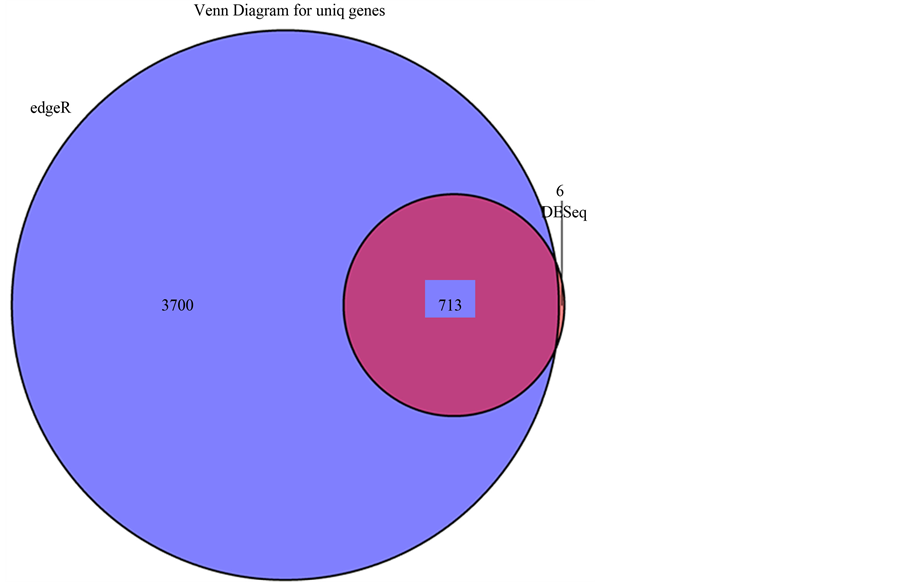 注:用DESeq和edgeR方法寻找差异基因的韦恩图。蓝色代表edgeR方法找出的特有基因,橘黄色为DESeq方法寻找出的特有基因,中间粉红色部分为两种方法共同鉴别出的差异基因。
注:用DESeq和edgeR方法寻找差异基因的韦恩图。蓝色代表edgeR方法找出的特有基因,橘黄色为DESeq方法寻找出的特有基因,中间粉红色部分为两种方法共同鉴别出的差异基因。
Figure 4. The Venn diagram for uniq genes
图4. 差异基因韦恩图
变化(后天改变)。目前TCGA已经确定将先研究肺癌、神经胶母细胞瘤和卵巢癌三种头号癌症和肿瘤的基因组图谱,从而了解整个TCGA项目的可行性,这项工作称为TCGA试验项目(该试验项目由美国全国癌症研究所和美国国家人类基因组研究所共同通过TCGA官方网站宣布)。TCGA试验项目的取得了里程碑式成果:1) 组织样本中确定独特的基因组变化[7] ;2) 根据特定的基因组改变和/或分子标记区分肿瘤亚型[8] ;3) 鉴定新的癌症相关的表观遗传学变化[9] ;4) 开发新的和改善原有的技术和分析工具[10] 。
近期,TCGA又在《Cell》公布了[8] 最新研究结果:对 12种不同癌症类型的3500个肿瘤样本的分子和遗传学特征进行生物信息学分析以确认相似的亚组(群),从而确定不同分子癌症亚型。这是一种完全区别于病理分类的肿瘤分子分类方法。另外在该项研究中,在一个亚型中的肿瘤差异或者不同癌症(来自于不同的器官)之间的重叠具有挑衅性。例如,经确认膀胱癌的至少三个亚型会出现不同的预后;这些亚型中的一个几乎与肺腺癌无法区分,而另一种与头颈肿瘤引起的鳞状细胞癌最为相似。这项研究强调并证实乳腺癌亚型之间存在差异,基底样乳腺癌,通常被称为三阴性乳腺癌,在分子水平上,比起乳腺癌的其他类型,这些基底样乳腺癌可能与卵巢癌和鳞状细胞癌起源的癌症有更多共同点,由此我们可以认为基底样乳腺癌实际上构成了自己的癌症类别。接着TCGA通过对599名患者的肿瘤样本和分子数据进行分析的结果绘制出了多形性胶质母细胞瘤(GBM)基因组景观图[11] 。这一更大的数据集及一种获得改进的分析算法使得能够更为细分基因扩增和缺失的信息比如7号染色体上的表皮生长因子受体(EGFR)扩增;鉴别了61个新的突变基因尤其是BRAF和FGFR有可能具有更直接的临床指导意义;确定了GBM四种亚型(神经型、原神经型、间质型和标准型)。
本研究以肝癌为例介绍了TCGA的基本情况包括数据处理、整合、数据水平及类型、统计分析方法,希望提供给广大科研工作者全面认识TCGA的机会。另外结合当下最热的生物信息学理论介绍了一种新的发现肿瘤差异基因包括mRNA、micRNA、拷贝数变异等,该方法相较于传统的芯片筛选具有样本数量大、费用小、分析简单等优势,为更多的人进行大规模的肝癌基因组学研究以及基于基因组学的后续功能研究提供了可能性。但是它也有自己的不足:免费版TCGA数据不包含患者基本情况及预后;只能描绘静态的突变或变异;不能反映基因水平到蛋白水平的改变。不管怎样TCGA项目将对癌症生物学、基因组学技术、生物储藏库和生物信息学领域的最新成果得到协调发展和最佳应用,科学合理的应用TCGA数据库可以使得科研工作尤其是肿瘤研究事半功倍。
基金项目
国家科技重大专项(2012ZX10002004)。
文章引用
贾俊君,何 宁,张 静,姜 骊,周燕飞,周 琳,郑树森, (2015) 基于TCGA数据库的肝癌发生关键基因筛选
Identification Key Genes of Hepatocellular Carcinoma Base on TCGA Database. 外科,01,1-8. doi: 10.12677/HJS.2015.41001
参考文献 (References)
- 1. Pang, R.W., Joh, J.W., Johnson, P.J., Monden, M., Pawlik, T.M., et al. (2008) Biology of hepatocellular carcinoma. Annals of Surgical Oncology, 15, 962-971.
- 2. He, J., Gu, D., Wu, X., Reynolds, K., Duan, X., et al. (2005) Major causes of death among men and women in China. New England Journal of Medicine, 353, 1124-1134.
- 3. Oshlack, A., Robinson, M.D. and Young, M.D. (2010) From RNA-seq reads to differential expression results. Genome Biology, 11, 220.
- 4. Venook, A.P., Papandreou, C., Furuse, J. and de Guevara, L.L. (2010) The incidence and epidemiology of hepatocellular carcinoma: A global and regional perspective. Oncologist, 15, 5-13.
- 5. Sanyal, A.J., Yoon, S.K. and Lencioni, R. (2010) The etiology of hepatocellular carcinoma and consequences for treatment. Oncologist, 15, 14-22.
- 6. Trevisani, F., Cantarini, M.C., Wands, J.R. and Bernardi, M. (2008) Recent advances in the natural history of hepatocellular carcinoma. Carcinogenesis, 29, 1299-1305.
- 7. Alexandrov, L.B., Nik-Zainal, S., Wedge, D.C., Aparicio, S.A., Behjati, S., et al. (2013) Signatures of mutational pro- cesses in human cancer. Nature, 500, 415-421.
- 8. Hoadley, K.A., Yau, C., Wolf, D.M., Cherniack, A.D., Tamborero, D., et al. (2014) Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell, 158, 929-944.
- 9. Barrio-Real, L., Benedetti, L.G., Engel, N., Tu, Y., Cho, S., et al. (2014) Subtype-specific overexpression of the Rac- GEF P-REX1 in breast cancer is associated with promoter hypomethylation. Breast Cancer Research, 16, 441.
- 10. Yang, D., Sun, Y., Hu, L., Zheng, H., Ji, P., et al. (2013) Integrated analyses identify a master microRNA regulatory network for the mesenchymal subtype in serous ovarian cancer. Cancer Cell, 23, 186-199.
- 11. Brennan, C.W., Verhaak, R.G., McKenna, A., Campos, B., Noushmehr, H., et al. (2013) The somatic genomic landscape of glioblastoma. Cell, 155, 462-477.