Asian Case Reports in Pediatrics
Vol.
10
No.
02
(
2022
), Article ID:
51773
,
9
pages
10.12677/ACRP.2022.102002
2岁男孩复发性腹股沟斜疝伴发育落后
罗统,田鑫*,王江,贺湘玲
湖南师范大学/湖南师范大学附属第一医院儿童医学中心/湖南省人民医院,湖南 长沙
收稿日期:2022年4月19日;录用日期:2022年5月19日;发布日期:2022年5月27日

摘要
患儿男,2岁11个月,复发性腹股沟疝1年余伴生长发育落后,体查生长发育落后,头大、矮小、手指短粗、皮肤黑、前额突出、弱视、肝大。实验室结果:心脏彩超示:心功能:EF:68%,FS:38%,二尖瓣、主动脉瓣瓣叶稍厚,主动脉瓣口前向流速稍快,二尖瓣、三尖瓣轻度反流,左室功能测值正常范围。腹部彩超示:肝大,副脾。裂隙灯检查见结膜无充血,角膜透明。IDS (Iduronate 2-Sulfatase)酶水平检测:0.8 nmol/h/ml。完善基因检测:发现IDS基因存在半合子突变,最终确诊为:1. 黏多糖贮积症II型;2. 左侧腹股沟斜疝。该病临床察觉较困难,早期易漏诊,该患儿起病至今历时2年余才确诊,因此当患儿出生后出现面部粗糙、发育迟缓、智力迟钝、生长迟缓、听力损失、心脏瓣膜增厚、肝脾肿大以及脐疝和腹股沟疝,需警惕黏多糖贮积症Ⅱ型,尽快完善尿GAGS,IDS酶来协助诊断,指导治疗,必要时完善基因检测以确诊。
关键词
黏多糖贮积症II型,IDS酶,尿糖胺聚糖(GAGS),复发性腹股沟疝,儿童

A 2-Year-Old Boy with Recurrent Inguinal Hernia and Developmental Delays
Tong Luo, Xin Tian*, Jiang Wang, Xiangling He
Hunan Normal University/Children’s Medical Center of First Affiliated Hospital of Hunan Normal University/Hunan Provincial People’s Hospital, Changsha Hunan
Received: Apr. 19th, 2022; accepted: May 19th, 2022; published: May 27th, 2022

ABSTRACT
A boy, 2 years old 11 months, was diagnosed with recurrent inguinal hernia for more than 1 year with growth and development backward, physical examination growth and development backward, head big, small, short thick fingers, dark skin, forehead prominent, amblyopia, large liver. Laboratory Results: Color Doppler showed echocardiography: cardiac function: EF: 68%, FS: 38%, mitral valve, aortic valve slightly thick, aortic valve anterior flow velocity slightly faster, mitral valve, tricuspid valve mild regurgitation, and left ventricular function measured normal range. Abdominal Color Doppler: large liver, accessory spleen. Slit-lamp examination showed no conjunctival congestion and corneal transparency. Detection of IDS Enzyme Level: 0.8 NMOL/H/ML. Perfect Gene Detection: Detection of IDS gene had a semi-zygote mutation. The final diagnosis was: 1. Mucopolysaccharidosis; 2. Left inguinal hernia. The disease is difficult to be detected clinically and easy to be missed in the early stage. It took more than 2 years for the child to be diagnosed. So when a child is born with facial roughness, stunted growth, mental retardation, growth retardation, hearing loss, heart valve thickening, hepatosplenomegaly, and umbilical and inguinal hernias, be on alert for mucopolysaccharidosis type II. Urinary GAGS and IDS should be conducted as soon as possible to help the diagnosis, guide treatment, and if necessary, improve gene detection to confirm the diagnosis.
Keywords:Mucopolysaccharidosis II, IDS Enzyme, Glycosaminoglycan (GAGS), Recurrent Inguinal Hernia, Child

Copyright © 2022 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/


1. 前言
黏多糖贮积症Ⅱ型由Charles Hunter博士在1917年首次报道,但因为认识不足,早期没有被认为是一种单独病种。黏多糖贮积症Ⅱ型是一种罕见的X连锁隐形遗传疾病,主要临床表现为:面部粗糙、发育迟缓、智力迟钝、生长迟缓、听力损失、心脏瓣膜增厚、肝脾肿大以及脐疝和腹股沟疝。由于黏多糖贮积症Ⅱ型的临床表现不具有特异性,在临床实践中容易漏诊、误诊。现对本院诊治的一例黏多糖贮积症Ⅱ型进行报道和回顾性分析,来提高临床医师对该病的认识。
2. 病例介绍
1) 病史:患儿男,2岁11个月,因“发现左侧阴囊可复性包块1年余,术后复发6月余”入院,患儿家属诉1年前无意中发现左侧阴囊肿大,约“核桃”大小,质软,无疼痛,站立、咳嗽或剧烈活动后出现,平卧后可自行消失。当时未予重视。近来发现患儿肿块较前明显增大,约“鸡蛋”大小,质软,无疼痛,站立、咳嗽或剧烈活动后出现,平卧后可自行消失。为求进一步治疗,今来我院门诊就诊,拟“左侧复发性腹股沟斜疝”收入小儿外科。
2) 个人史、既往史、家族史:患儿系第一胎第一产,出生时巨头畸形,既往2018年1月、2018年7月在株洲市中心医院诊断为“左侧腹股沟疝”,行传统疝囊高位结扎术。语言功能较同龄人稍有落后,无过敏体质。
3) 入院查体:体温:36.4℃,脉搏:95次/分,呼吸:16次/分,体重:16 Kg。患儿语言功能较同龄人稍落后,头围52 cm,矮小,手指短粗,皮肤黑,前额突出、弱视。双肺呼吸音清,均未闻及干湿性啰音。腹软,全腹无压痛及反跳痛,肝肋下3~4 cm,质软,脾未触及。专科检查:站立时左侧腹股沟区可及4 cm × 4 cm大小包块,质软,坠入阴囊,直立位出现,平卧后自行还纳入腹腔,随咳嗽而增大,压住内环口后肿物不出现,松开后出现。右侧腹股沟区未及明确包块,阴囊透光实验阴性。
4) 辅助检查:血常规(2020-10-26)血常规示:白细胞计数6.39 (×109/L),血红蛋白125 g/L,中性粒细胞百分率30.8(%)↓,淋巴细胞百分率57.9(%)↑;血栓止血:定量纤维蛋白原1.63↓,活化部份凝血活酶时间36.1↑,APPT_比值1.34↑,凝血酶时间24.4↑,纤维蛋白(原)降解产物7.1↑;生化报告(日立008):谷草转氨酶47.3(U/L)↑,碱性磷酸酶214.4(U/L)↑;血型 + 抗筛:ABO正定型B型,Rh(D)阳性↑;输血前四项、微量元素检测、C-反应蛋白、降钙素原正常。心电图示(electrocardiogram图1):窦性心动过速。肺功能正常。胸片示(Chest X-ray图2):双肺未见实变,心膈正常。双侧腹股沟彩超示(Superficial organ ultrasound图3):左侧腹股沟区包块,考虑疝。

Figure 1. Electrocardiogram
图1. 心电图

Figure 2. Chest X-ray
图2. 胸部X线

Figure 3. Superficial organ ultrasound
图3. 浅表器官超声
3. 诊断思维
患儿病例特点:1) 学龄前男孩,11个月大时发现左侧腹股沟疝;2) 主要表现为腹股沟疝复发;3) 头围大,矮小,手指短粗,皮肤黑,前额突出,弱视,肝大;4) 语言功能较同龄人稍落后;5) 双侧腹股沟彩超示:左侧腹股沟区包块。
该患儿临床表现主要为腹股沟疝复发,复发原因主要从以下几个方面考虑,1) 手术时机把握不恰当:患儿11个月大时诊断为左侧腹股沟疝,在外院行传统疝囊高位结扎术,手术是儿童腹股沟疝的唯一根治手段,有专家认为手术时机的选择在出生后1岁至2岁更为合适 [1],儿童腹股沟疝多为先天性腹壁结构发育不良,1岁前患儿腹壁和腹膜发育基础情况较差,同时男性患儿的年龄 < 1岁也是疝气复发的独立危险因素 [2],且患儿经常哭闹,消化不良等因素还会引起腹腔内压力增高,这些均会增加术后疝气复发的几率。2) 不同手术方式的选择:患儿在外院行传统疝囊高位结扎术,而有研究表明开放性修复腹股沟疝的术式同LPEC术相比,有更高的复发率,并发症发生率也更高 [3],对于复发性疝如果之前做的是传统开放式手术,复发后建议予以LPEC术,反之亦然 [4]。3) 围手术期腹股沟疝复发的危险因素中包括了外科手术医师的经验及技术水平,LPEC平均学习曲线约为30例左右,传统开放手术平均学习曲线更长一些,且外科医师有限手术经验属于高水平证据的危险因素 [5]。4) 其他可引起腹压增高全身系统疾病:① 肝豆状核变性?:患儿有肝大,语言功能较同龄人落后但无运动障碍、肢体震颤等表现,眼部未见角膜色素环。故可能性不大,完善血清铜蓝蛋白检查协诊。② 地中海贫血?患儿有肝大,但无贫血、黄疸等表现,故可能性不大。③ 黏多糖贮积症(Mucopolysaccharidosis)?:患儿喜憋气,左侧腹股沟疝多次复发,查体前额突出、头大(头围52 cm)、皮肤黝黑、手指粗短,生长发育较同龄儿稍有落后(语言功能较同龄人稍落后),故需考虑该病。需完善尿糖胺聚糖(GAGs)水平检测、IDS酶水平、听力、视力、心脏彩超等相关检查,必要时完善黏多糖特异性基因IDS检测以协助诊断。
4. 进一步检查
心脏彩超示(Cardiac ultrasound,图4):心功能:EF:68%,FS:38%,二尖瓣、主动脉瓣瓣叶稍厚,主动脉瓣口前向流速稍快,二尖瓣、三尖瓣轻度反流,左室功能测值正常范围。腹部彩超示(Abdominal ultrasound,图5):肝大,副脾。裂隙灯检查见结膜无充血,角膜透明。IDS酶水平检测:0.8 nmol/h/ml。血清铜蓝蛋白:305 mg/L。
IDS对于诊断黏多糖贮积症具有很高的敏感性及特异性,取得患儿家属同意后,取患儿及其父母外周血,提取DNA,进行基因分析,检测方法:本人–高通量测序,父亲–Sanger验证,母亲–Sanger验证;发现chrX:1485 79660染色上IDS基因有1个半合子突变:1) c.686A > G (exon5, NM_000202),导致氨基酸改变p.H229R,为错义突变。根据ACMG指南,该变异初步判定为致病性变异(Pathogenic)PS4 + PM1 + PM2 + PM5 + PP3:PS4:文献数据库已有该位点(Mucopolysaccharidosis II)的病例报道,变异标签为DM (致病突变),ClinVar数据库无该位点致病性分析结果;PM1:该变异位于突变热点区域;PM2:在正常人群数据库中的频率为−,为低频变异;PM5:相同位置的突变在文献数据库已有Mucopolysaccharidosis II报道但氨基酸变化不同,文献报道的是c.685C > T;p.H229Y,变异标签为DM (致病突变) Jonsson, et al. Am J Hum Genet, 56,597,1995[PMID:7887413]|27695081;PP3:生物信息学蛋白功能综合性预测软件REVEL预测结果为(有害),SIFT、PolyPhen_2、MutationTaster、GERP+预测结果分别为有害、有害、有害、有害;经家系验证分析,c.686A > G (p.H229R)受检人之父该位点无变异,受检人之母该位点杂合变异。(图6)
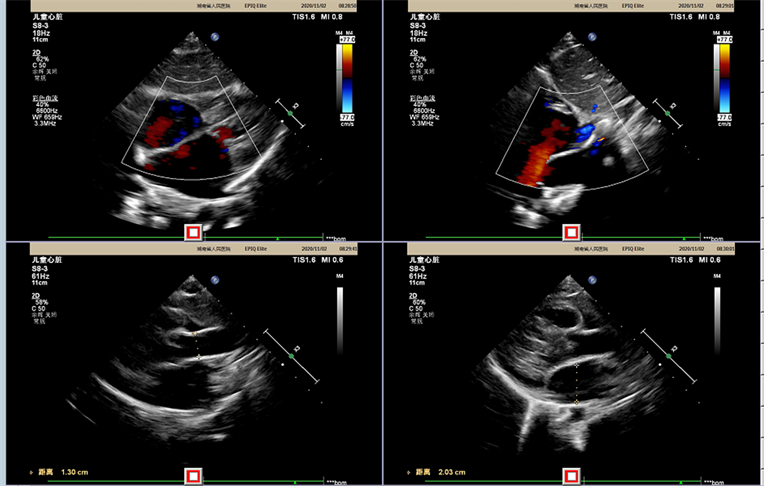
Figure 4. Cardiac ultrasound
图4. 心脏彩超

Figure 5. Abdominal ultrasound
图5. 腹部超声
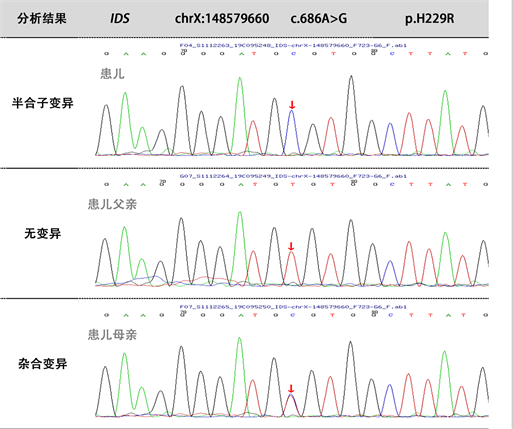
Figure 6. Sanger sequence map of IDS gene in children and their parents. There was a semi-zygote mutation in the children’s IDS gene, C.686A > G (p.H229R) had no change in the father, and the mother had a heterozygous mutation in the site. The mutation point is located at the Arrow
图6. 患儿及父母IDS基因Sanger测序图患儿IDS基因存在1个半合子突变,c.686A > G (p.H229R)患儿父亲该位点无变异,患儿母亲该位点杂合突变。突变点位于箭头所示
5. 诊断及依据
诊断:1黏多糖贮积症Ⅱ型2左侧复发性腹股沟斜疝。依据:1) 男性,婴幼儿,生长发育落后,头围大,矮小,手指短粗,皮肤黑,前额突出、弱视,肝大,伴反复腹股沟疝;2) 心脏彩超:二尖瓣、主动脉瓣瓣叶稍厚;3) IDS酶水平检测:0.8 nmol/h/m,IDS基因存在半合子突变,且突变遗传自患儿母亲。2.左侧复发性腹股沟斜疝:1) 专科检查:站立时左侧腹股沟区可及4 cm × 4 cm大小包块,质软,坠入阴囊,直立位出现,平卧后自行还纳入腹腔,随咳嗽而增大,压住内环口后肿物不出现,松开后出现。2) 双侧腹股沟彩超示:左侧腹股沟区包块。
6. 临床经过
入院完善相关术前检查后于2020-10-28在全麻下行腹腔镜左侧疝囊高位结扎术。手术顺利。术后予以伤口加压、垫高阴囊、营养补液等对症治疗,患者病情好转,伤口愈合良好。随访1年斜疝未复发。确诊后建议行酶替代及造血干细胞移植治疗,但由于经济原因,暂未实施。临床继续随访。
7. 讨论
黏多糖贮积病是一组遗传性疾病 [6],因为参与黏多糖(又称糖胺聚糖GAGs)代谢的溶酶体酶缺失而引起个体发育异常以及代谢产物贮积在各组织器官造成的功能障碍。分类包括I型(Hurler综合征,Hurler-Scheie和Scheie综合征),II型(Hunter综合征),III型(Sanfilippo综合征),IV型(Morquio综合征)和VI型(Maroteaux-Lamy综合征) [7]。发病机制:哺乳动物组织中皆存在糖胺聚糖 [8],糖胺聚糖在细胞中发挥各种重要作用,包括组成细胞外基质与膜、参与细胞水合作用、更是在细胞信号转导中起着关键作用 [9]。而糖胺聚糖在体内的清除是通过溶解酶对其的序列特异性降解进行的 [10]。所以降解步骤中的任何一环缺失都会引起糖胺聚糖组成成分的改变(包括硫酸乙酰肝素的增加,硫酸角蛋白的减少)以及底物的贮积(包括GM2和GM3神经节苷脂和乳糖基神经酰胺) [11],进而引发神经与骨骼等各系统的生长发育异常和功能障碍。腹股沟疝的发生与多种因素有关,其中一个原因是腹股沟管内结缔组织异常,腹股沟管由胶原蛋白、弹性蛋白和微纤维组成的弹性纤维以及细胞外基质的糖胺聚糖等成分组成,影响这些成分的每一种遗传异常都会增加腹股沟疝的发病率 [12]。有研究表明在复发性疝病、成人腹股沟疝及切口疝中,间质胶原蛋白的丰度与组成发生了显著的变化,这些证据提示了疝的形成与复发可能与组织更新紊乱有关 [13]。而糖苷聚糖成分变化对胶原蛋白及其他相关成分合成的影响在疝形成的过程中发挥着作用 [14]。
本例患儿因反复腹股沟斜疝伴生长发育落后入院,被确诊为黏多糖贮积症Ⅱ型。黏多糖贮积症Ⅱ型典型表现包括:面部粗糙、发育迟缓、智力迟钝、生长迟缓、听力损失、心脏瓣膜增厚、肝脾肿大以及脐疝和腹股沟疝,这些症状在患儿2岁至4岁时变得明显 [15]。
目前对于黏多糖贮积病Ⅱ型的治疗主要为酶代替治疗(Enzyme Replacement Therapy, ERT) [16],中国专家共识推荐患儿在确诊后即可开始ERT,早期进行ERT可减缓病程进展,改善预后。治疗剂量:推荐剂量为0.5 mg/kg体重,每周1次静脉输注,每次输注时间宜控制在1-3h内,如发生输液相关反应,可延长输注时间,一般不应超过8 h。伴有严重认知障碍的重型患儿,若预期ERT可改善躯体症状,亦可以考虑接受ERT。在伴有威胁生命的合并症;已发展至疾病晚期,ERT治疗预计不会获益 [17]。除了酶代替治疗,还可以考虑异基因造血干细胞移植,移植后由健康供者巨噬细胞透过血脑屏障后转换为脑小胶质细胞,脑小胶质细胞能够分泌有活性的酶,来改善患者部分中枢神经系统症状 [18]。相较于ERT,它的优势为:可跨越血脑屏障,改善中枢神经系统症状;一次性治疗,费用较ERT低,但存在移植相关风险性。其他治疗包括:1) 细胞治疗:新鲜血液的输注;腹膜内植入携带过表达IDS的C2C12鼠成肌细胞的藻酸盐微胶囊;2) 底物减少疗法:木黄酮可诱导MPS1型和2型3A3B型成纤维细胞中的GAG的减少,且木黄酮可透过血脑屏障,存在与ERT联合治疗的潜力,3) 基因治疗:利用腺病毒、慢病毒等载体将IDS基因带入体内,可提高脑组织中IDS的活性。并发症的管理:压迫相关症状:交通性脑积水、髓压迫及腕管综合征症状的出现,需完善减压手术来治疗,癫痫的发作,通常使用抗惊厥药物 [19];其他并发症均以对症治疗为主。
8. 结语
黏多糖贮积症Ⅱ型是临床罕见的遗传性X隐形遗传疾病,由IDS基因突变导致的黏多糖及其代谢产物在各组织上沉积引起生长发育落后、器官功能障碍。通常表现为:面部粗糙、发育迟缓、智力迟钝、生长迟缓、听力损失、心脏瓣膜增厚、肝脾肿大以及脐疝和腹股沟疝。在面对有上述症状的婴幼儿,我们需要考虑到黏多糖贮积症Ⅱ型的可能性,完善尿GAGS水平检查,IDS酶水平测定,同时完善基因检测以尽早明确诊断。对于有家庭遗传史的家庭建议完善产前诊断,达到早期诊断,早期干预的目的。
基金项目
该病例报道已获得患方知情同意且符合相关伦理要求,感谢患方提供的宝贵临床资料,感谢湖南省自然科学基金青年项目(2021JJ40295)资助。
文章引用
罗 统,田 鑫,王 江,贺湘玲. 2岁男孩复发性腹股沟斜疝伴发育落后
A 2-Year-Old Boy with Recurrent Inguinal Hernia and Developmental Delays[J]. 亚洲儿科病例研究, 2022, 10(02): 7-15. https://doi.org/10.12677/ACRP.2022.102002
参考文献
- 1. 陈辉球, 文友良, 李斐. 小儿腹股沟斜疝术后复发原因分析[J]. 健康必读, 2019(4): 288-289.
- 2. 王江等. 腹股沟疝患儿腹腔镜经皮腹膜外闭合术后疝复发及对侧腹股沟疝发生的危险因素分析[J]. 中国现代医学杂志, 2021, 31(11): 75-80. https://doi.org/10.3969/j.issn.1005-8982
- 3. Miyake, H., et al. (2016) Comparison of Percutane-ous Extraperitoneal Closure (LPEC) and Open Repair for Pediatric Inguinal Hernia: Experience of a Single Institution with over 1000 Cases. Surgical Endoscopy, 30, 1466-1472. https://doi.org/10.1007/s00464-015-4354-z
- 4. Berndsen, M.R., Gudbjartsson, T. and Berndsen, F.H. (2019) [Inguinal Hernia—Review]. Laeknabladid, 105, 385-391. https://doi.org/10.17992/lbl.2019.09.247
- 5. HerniaSurge Group (2018) International Guidelines for Groin Hernia Management. Hernia: The Journal of Hernias and Abdominal Wall Surgery, 22, 1-165. https://doi.org/10.1007/s10029-017-1668-x
- 6. Fitzgerald, J. and Verveniotis, S.J. (1998) Morquio’s Syndrome. A Case Report and Review of Clinical Findings. New York State Dental Journal, 64, 48-50.
- 7. Silveira, M., et al. (2018) Audiometric Evaluation in Individuals with Mucopolysaccharidosis. Clinics (Sao Paulo), 73, e523. https://doi.org/10.6061/clinics/2018/e523
- 8. Fecarotta, S., et al. (2020) Pathogenesis of Mucopolysaccharidoses, an Update. International Journal of Molecular Sciences, 21, Article 2515. https://doi.org/10.3390/ijms21072515
- 9. Prydz, K. (2015) Determinants of Glycosaminoglycan (GAG) Structure. Biomolecules, 5, 2003-2022. https://doi.org/10.3390/biom5032003
- 10. Raman, R., Sasisekharan, V. and Sasisekharan, R. (2005) Structural In-sights into Biological Roles of Protein-Glyco- saminoglycan Interactions. Cell Chemical Biology, 12, 267-277. https://doi.org/10.1016/j.chembiol.2004.11.020
- 11. Ernst, S., et al. (1995) Enzymatic Degradation of Glycosa-minoglycans. Critical Reviews in Biochemistry and Molecular Biology, 30, 387-444. https://doi.org/10.3109/10409239509083490
- 12. Barnett, C., et al. (2009) Looking Past the Lump: Genetic As-pects of Inguinal Hernia in Children. Journal of Pediatric Surgery, 44, 1423-1431. https://doi.org/10.1016/j.jpedsurg.2008.12.022
- 13. Klinge, U., Binnebösel, M. and Mertens, P.R. (2006) Are Col-lagens the Culprits in the Development of Incisional and Inguinal Hernia Disease? Hernia, 10, 472-477. https://doi.org/10.1007/s10029-006-0145-8
- 14. Martin, C.L., et al. (2020) A Role for Monosaccharides in Nucle-ation Inhibition and Transport of Collagen. Bioelectricity, 2, 186-197. https://doi.org/10.1089/bioe.2020.0013
- 15. Khan, S.A., et al. (2017) Epidemiology of Mucopolysaccharidoses. Molecular Genetics and Metabolism, 121, 227-240. https://doi.org/10.1016/j.ymgme.2017.05.016
- 16. Kim, C., et al. (2017) Comparative Study of Idursulfase Beta and Idursulfase in Vitro and in Vivo. Journal of Human Genetics, 62, 167-174. https://doi.org/10.1038/jhg.2016.133
- 17. 中华医学会儿科学分会内分泌遗传代谢学组. 黏多糖贮积症II型临床诊断与治疗专家共识[J]. 中华儿科杂志, 2021, 59(6): 446-451.
- 18. Araya, K., et al. (2009) Localized Donor Cells in Brain of a Hunter Disease Patient after Cord Blood Stem Cell Transplantation. Molecular Genetics and Metabo-lism, 98, 255-263. https://doi.org/10.1016/j.ymgme.2009.05.006
- 19. Muenzer, J., et al. (2009) Multidisciplinary Management of Hunter Syndrome. Pediatrics, 124, e1228-e1239. https://doi.org/10.1542/peds.2008-0999