Journal of Organic Chemistry Research
Vol.
10
No.
04
(
2022
), Article ID:
58438
,
8
pages
10.12677/JOCR.2022.104012
无金属介入的去芳香化反应研究 进展
李瑞琦
浙江师范大学化学与生命科学学院,浙江 金华
收稿日期:2022年8月29日;录用日期:2022年11月18日;发布日期:2022年11月28日

摘要
一些易得的芳香族原料和复杂大分子的中间体可以通过去芳香化反应合成,因此在过去十年中,去芳香化反应的迅速发展为天然产物提供了新的合成方法。本综述将重点介绍非金属催化条件下去芳香化反应的研究进展以及各类策略在合成应用方面的进展,并将去芳香化反应分为以下几类,包括氧化还原型去芳香化反应、亲核亲电型去芳香化反应和环加成型去芳香化反应。
关键词
芳烃,去芳香化,无金属介入

Research Progress of Dearomatization Reactions without Metal Intervention
Ruiqi Li
City Province College of Chemistry and Life Science, Zhejiang Normal University, Jinhua Zhejiang
Received: Aug. 29th, 2022; accepted: Nov. 18th, 2022; published: Nov. 28th, 2022

ABSTRACT
Some readily available aromatic feedstocks and intermediates of complex macromolecules can be synthesized by dearomatization reactions, so the rapid development of dearomatization reactions in the last decade has provided new synthesis methods for natural products. This review will focus on the research progress of dearomatization reactions under non-metallic catalytic conditions and the progress of various strategies in synthetic applications and will classify dearomatization reactions into the following categories, including REDOX dearomatization reactions, nucleophilic and electrophilic dearomatization reactions and cycloadditive dearomatization reactions.
Keywords:Arene, Dearomatization Reactions, Metal Free Reaction

Copyright © 2022 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/


1. 引言
芳香族分子属于最基本最丰富的有机化合物类别之一,广泛应用于分子科学的各个领域。去芳香反应能够干扰芳香族π-体系并提供不饱和的官能化的产物,因而近几年成为了有些化学家研究的热点领域。芳烃作为起始原料,广泛的碳氢化合物来源使得去芳香化反应可以快速获得更复杂和多功能性的中间体,这种由简单到复杂的直接合成逻辑在天然产物合成小分子药物化学领域越来越流行,去芳香化策略的在天然产物中的应用实例更加丰富 [1]。
原则上,每种芳香族化合物都可以去芳香化,然而由于芳烃的共振稳定性较高,杂环芳烃(如呋喃、吡咯或吡啶)比芳烃(如苯)更容易发生去芳香化过程 [2]。对于芳烃,去芳香化反应可以分为几类,包括氧化还原反应、亲核亲电反应和环加成反应。本综述的目的是总结最近以上几类反应的发展,特别是在该反应在天然产物中的合成应用。
2. 氧化型去芳香化反应
在许多情况下,芳烃的氧化去芳香化伴随着杂原子的引入,特别是醇、羧酸和酰胺。在氧化苯酚中添加醇可方便地获得1,3-和1,4-环己二烯酮。中间体(见图1,式1)的确切结构随氧化剂的变化而改变,亲核加成的位置可能受到氧化剂的影响,但加成通常发生在具有最大LUMO系数的二烯基碳上。通过该过程产生的邻位和对位醌大部分都非常活泼,尤其是1,3-型产物(见图1,式2),表现出通过4+2环加成自发二聚的趋势 [3]。

Figure 1. Overview of oxidative dearomatization pathways
图1. 氧化去芳香化途径概述
Gaunt等人 [4] 开发了一种氧化去芳香化后有机催化分子内的Michael加成,可以对位取代苯酚构建成为含对映体的双环结构。此外还报道了4,4-二取代环己二烯酮通过有机催化的Stetter反应和含手性氢键的尿素类催化剂催化的Michael加成反应类似的不对称反应(见图2) [5] [6]。

Figure 2. Desymmetrization of a p-quinol obtained from oxidative dearomatization
图2. 通过氧化去芳香化获得的不对称的醌醇
烯丙基硅烷是一种碳亲核试剂,与高价碘试剂诱导的氧化去芳香化反应结合表现良好 [7]。2007年Nicolaou等人 [8] 曾报道过分子间和分子内的烯丙基硅烷添加到酚鎓阳离子中的反应,后一种反应类型可以合成强效抗生素的中间体(−)-钝顶霉素(见图3,式9) [8]。

Figure 3. Oxidative dearomatization route to (-)-platensimycin
图3. 氧化去芳香化途径合成(-)-钝顶霉素
3. 亲核和亲电型去芳香化反应
3.1. 亲电型去芳香化反应
酚具有烯醇互变异构体的稳定形式,酚酸根阴离子也具有双重亲核性。因此,酚酸根阴离子与适当的有机亲电试剂(例如烷基卤化物和磺酸盐)反应成键,键的形成发生在取代的芳烃碳上时,使得稳定的环己二烯酮产物的分离,并完成烷基化的去芳香化(见图4) [9]。
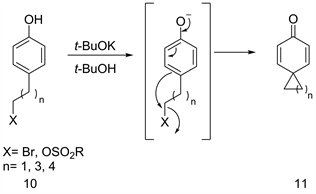
Figure 4. Intramolecular alkylative dearomatization
图4. 分子内烷基化的去芳香化反应
分子内烷基化去芳香化为构建螺环己二烯酮环是非常方便的方法。该反应已用于许多全合成,目标是构建天然产物中发现的萜类环系统,如雪松烯、海因索尔、考脲、β-维替酮和异长叶烯 [10]。随后,烷基化去芳香化是合成更复杂天然产物中的关键步骤,如杜卡霉素SA [11]、可的松A [12] 和普坦霉素(见图5,式13) [13]。值得注意的是,因为它说明在烷基化反应中使用硅烷基可以保护酚阴离子。

Figure 5. Approach to the core structure of platensimycin
图5. 平板霉素核心结构的合成
2019年,Peng等人 [14] 描述了芳基碘的去芳构化通过一个前所未有的“重排/加成”序列(见图6)。这个过程包括两个阶段。芳基碘与α-苯乙烯腈快速[3,3] sigma重排,得到高度亲电的去芳构中间体。该反应不仅破坏了芳基碘的芳香性,而且顺序地组装了两个不同的官能团,从而得到多取代的脂环产物。后续他们用高价硫做为亲核物种也得到此类产物 [15]。
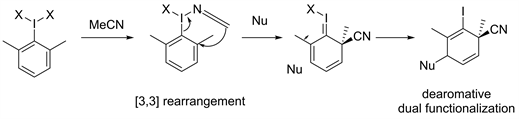
Figure 6. Dearomative Dual Functionalization of Aryl Iodanes
图6. 芳基碘烷的去芳香双官能团化
3.2. 亲核型去芳香化反应
在过去几十年中,已经有许多关于活性物质亲核进攻芳香环引发的去芳香化反应的报道。比如,乙烯基碘分子内去芳香化反应,相应的丁二烯基碘化物在形成乙烯基锂后,在-78℃下加入亲核试剂淬灭反应后分离出去芳香化产物(见图7,式15a-15f) [16] [17]。

Figure 7. Intramolecular nucleophilic dearomatization of butadieny-lithium naphthalene and derivatives
图7. 丁二烯基萘锂及其衍生物的分子内亲核去芳香化
当以苯基丁二烯为底物时,可以获得1,2-和1,4-两种去芳香化产物 [17]。异构体产物比例由所用的芳香底物和亲电试剂决定。苯甲醛作为亲核试剂淬灭反应时,分离得到1,4-去芳香化化合物为主要产物(见图8,式18b),当使用取代苯甲醛作为亲核试剂时,1,2-去芳香化产物作为主要产物(见图8,式18a)。酮和亚胺作为亲核试剂时,1,4-去芳香化化合物是唯一的产物。基于这些结果,作者认为反应的选择性取决于芳香底物和亲核试剂的空间位阻而不是电子效应。

Figure 8. Intramolecular nucleophilic dearomatization of butadieny-lithium benzene and derivatives
图8. 丁烯基苯锂及其衍生物的分子内亲核脱芳去芳香化
将锂化的亲电试剂添加到苯衍生物中也可用于贝类毒素的全合成,包括异多米酸B (见图9,式21)。芳基酰胺脱质子使分子内亲核进攻苯环,分离得到不饱和酮(见图9,式20) [18]。
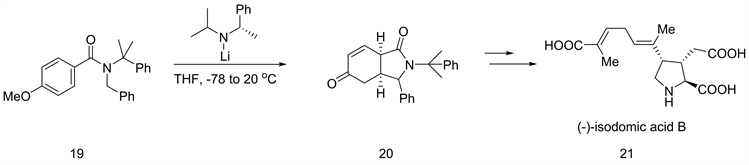
Figure 9. Enantioselective synthesis of isodomic acid B
图9. 异多米酸B的对映选择性合成
4. 环加成型去芳香化反应
加成反应能够直接引入各种官能团,已知芳香烃和烯烃之间的环加成过程通过形成桥连或稠合的双环去芳香化产物来增加结构复杂性。[2 + 2]、[3 + 2]和[4 + 2]环加成都已在文献中报道,在光化学体系下,芳烃和烯烃两者的激发态的加成,邻、间或对位选择性是由一方的固有电子性质决定。烯烃和芳烃之间环加成是合成一系列多环天然产物的关键策略,这一关键步骤通常发生在合成路线的早期。
两种令人印象深刻的合成实例,烯烃(见图10,式22和式25)进行分子内1,3-加成以形成环丙烷中间体(见图10,式23和式26),这两种中间体后续几步可以合成的天然产物Penifulvin C (见图10,式24) [19] 和Ceratopicanol (见图10,式27) [20],这种间烯烃-芳烃环加成能够快速获得复杂的中间体。

Figure 10. Alkene-arene dearomative cycloaddition and applications of meta-alkene-arene cycloaddition in total synthesis
图10. 烯烃和芳烃去芳香化加成反应及其在全合成中的应用
除了烯烃和芳烃之间的环加成反应外,早在1982年,在苯胺的情况下(Himbert芳烃环加成)就建立了烯和芳烃之间分子内的Diels-Alder反应 [21]。这种令人惊讶的反应性归因于氮取代基的活化和烯的高能量。后续的改进是用连烯取代酰胺上的氮,由于这类环加成反应几乎是热中性的,多种烯烃底物可转化为产物(见图11,29a-29d) [22]。

Figure 11. Arene-allene cycloaddition
图11. 芳烃-丙烯环加成反应
虽然1971年Huisgen等人 [23] 首次报道了1,3-偶极环加成反应,但这这种去芳香化反应到2011年才有进一步的发展。吸电子芳烃和硅烷化的胺和三氟乙酸原位生成的富电子、不稳定的甲亚胺叶立德之间的分子间反应,得到单环或双环化合物(见图12,31a-31f) [24] [25]。该反应的区域选择性高度依赖于底物,得到相应的产物(如图12,31a-31c)。对于萘硝基芳烃图12 (31d-31f),单环加合物的产率很高,可能是由于苯乙烯的稳定性。
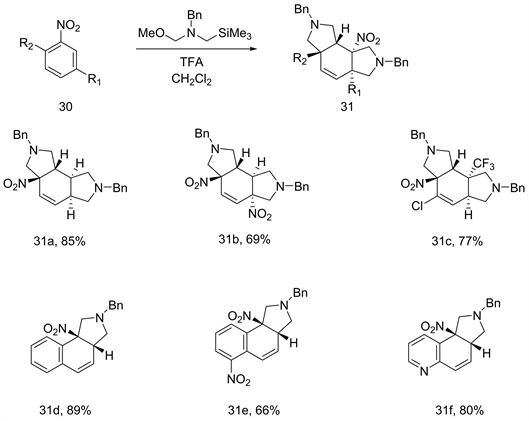
Figure 12. [3 + 2] cycloaddition between azomethane ylide and nitroarenes
图12. 偶氮甲烷叶立德和硝基芳烃之间的[3 + 2]环加成反应
5. 总结
近年来该领域得到了显著的发展和完善,去芳香化的概念已成为有机合成中一种重要而有力的策略。现成的芳烃底物合成结构复杂的脂环系统可以由去芳香化过程得到。与烯烃化学相反,去芳构化反应中的官能团兼容性和位点选择性仍然非常有限,不适合用于更复杂的分子环境中进行去芳香化反应及其后期的应用。然而,鉴于芳烃在分子科学中的丰富性和高频率使用,去芳香化反应领域会继续发展。
文章引用
李瑞琦. 无金属介入的去芳香化反应研究进展
Research Progress of Dearomatization Reactions without Metal Intervention[J]. 有机化学研究, 2022, 10(04): 119-126. https://doi.org/10.12677/JOCR.2022.104012
参考文献
- 1. Roche, S.P. and Porco Jr., J.A. (2011) Dearomatization Strategies in the Synthesis of Complex Natural Products. Angewandte Chemie International Edition, 50, 4068-4093. https://doi.org/10.1002/anie.201006017
- 2. Zhuo, C.-X., Zhang, W. and You, S.-L. (2012) Catalytic Asymmetric Dearomatization Reactions. Angewandte Chemie International Edition, 51, 12662-12686. https://doi.org/10.1002/anie.201204822
- 3. Magdziak, D., Meek, S.J. and Pettus, T.R.R. (2004) Cyclohexadienone Ketals and Quinols: Four Building Blocks Potentially Useful for Enantioselective Synthesis. Chemical Reviews, 104, 1383-1429. https://doi.org/10.1021/cr0306900
- 4. Vo, N.T., Pace, R.D.M., O’Hara, F. and Gaunt, M.J. (2008) An Enantioselective Organocatalytic Oxidative Dearomatization Strategy. Journal of the American Chemical Society, 130, 404-405. https://doi.org/10.1021/ja077457u
- 5. Liu, Q. and Rovis, T. (2006) Asymmetric Synthesis of Hydrobenzofuranones via Desymmetrization of Cyclohexadienones Using the Intramolecular Stetter Reaction. Journal of the American Chemical Society, 128, 2552-2553. https://doi.org/10.1021/ja058337u
- 6. Gu, Q. and You, S.-L. (2011) Desymmetrization of Cyclohexadienones via Asymmetric Michael Reaction Catalyzed by Cinchonine-Derived Urea. Organic Letters, 13, 5192-5195. https://doi.org/10.1021/ol202073p
- 7. Zheng, C., Wang, L., Li, W., Wang, L. and Wang, D.Z. (2013) Ortho-Dearomatization of Phenols Creating All-Carbon Spiro-Bicycles. Organic Letters, 15, 4046-4049. https://doi.org/10.1021/ol401863k
- 8. Nicolaou, K.C., Edmonds, D.J., Li, A. and Tria, G.S. (2007) Asymmetric Total Syntheses of Platensimycin. Angewandte Chemie International Edition in English, 46, 3942-3945. https://doi.org/10.1002/anie.200700586
- 9. Murphy, W.S. and Wattanasin, S. (1983) Anionic Cyclization of Phenols. Chemical Society Reviews, 12, 213-250. https://doi.org/10.1039/cs9831200213
- 10. Kraus, G.A. and Kesavan, S. (2005) Preparation of Advanced Intermediates for the Synthesis of both Methyllycaconitine and Racemulsonine via a Common Intermediate. Tetrahedron Letters, 46, 1111-1113. https://doi.org/10.1016/j.tetlet.2004.12.092
- 11. Boger, D.L. and Machiya, K. (1992) Total Synthesis of (+)-Duocarmycin SA. Journal of the American Chemical Society, 114, 10056-10058. https://doi.org/10.1021/ja00051a045
- 12. Wang, Z., Dai, M., Park, P.K. and Danishefsky, S.J. (2011) Synthetic Studies toward (+)-Cortistatin A. Tetrahedron, 67, 10249-10260. https://doi.org/10.1016/j.tet.2011.10.026
- 13. McGrath, N.A., Bartlett, E.S., Sittihan, S. and Njardarson, J.T. (2009) A Concise Ring-Expansion Route to the Compact Core of Platensimycin. Angewandte Chemie International Edition, 48, 8543-8546. https://doi.org/10.1002/anie.200903347
- 14. Zhao, W.Z. and Huang, X. (2019) Dearomative Dual Functionalization of Aryl Iodanes. Angewandte Chemie International Edition, 58, 17210-17214. https://doi.org/10.1002/anie.201909019
- 15. Huang, X., Zhang, Y.G. and Peng, B. (2020) Dearomatization of Aryl Sulfoxides: A Switch between Mono- and Dual-Difluoroalkylation. Chemical Science, 11, 3048-3053. https://doi.org/10.1039/D0SC00244E
- 16. Wang, Z. and Xi, Z. (2006) Dearomatizing Anionic Cyclization of 1-Lithio-4-naphthyl-1,3-butadienes Leading to the Formation of Spiro Cyclopentadiene Derivatives. Synlett, No. 8, 1275-1277. https://doi.org/10.1055/s-2006-939083
- 17. Liu, L., Wang, Z., Zhao, F. and Xi, Z. (2007) Dearomatizing Anionic Cyclization and Novel Skeletal Rearrangement: High Yield Formation of Multiply Substituted Bicyclic or Polycyclic Spirocyclopentadienes and Phenanthrene Derivatives from 4-Aryl 1-Lithio-1,3-butadienes. The Journal of Organic Chemistry, 72, 3484-3491. https://doi.org/10.1021/jo070160u
- 18. Lemiere, G., Sedehizadeh, S., Toueg, J., Fleary-Roberts, N. and Clayden, J. (2011) A General Synthetic Approach to the Amnesic Shellfish Toxins: Total Synthesis of (−)-Isodomoic Acid B, (−)-Isodomoic Acid E and (−)-Isodomoic Acid F. Chemical Communications, 47, 3745-3747. https://doi.org/10.1039/c1cc00048a
- 19. Gaich, T. and Mulzer, J. (2010) From Silphinenes to Penifulvins: A Biomimetic Approach to Penifulvins B and C. Organic Letters, 12, 272-275. https://doi.org/10.1021/ol902594b
- 20. Baralotto, C., Chanon, M. and Julliard, M. (1996) Total Synthesis of the Tricyclic Sesquiterpene (±)-Ceratopicanol. An Illustration of the Holosynthon Concept. The Journal of Organic Chemistry, 61, 3576-3577. https://doi.org/10.1021/jo960382k
- 21. Himbert, G. and Henn, L. (1982) Intramolecular Diels-Alder Reaction of Allenecarboxanilides. Angewandte Chemie International Edition in English, 21, 620. https://doi.org/10.1002/anie.198206201
- 22. Schmidt, Y., Lam, J.K., Pham, H.V., Houk, K.N. and Vanderwal, C.D. (2013) Studies on the Himbert Intramolecular Arene/Allene Diels-Alder Cycloaddition. Mechanistic Studies and Expansion of Scope to All-Carbon Tethers. Journal of the American Chemical Society, 135, 7339-7348. https://doi.org/10.1021/ja4025963
- 23. Huisgen, R. and Scheer, W. (1971) Dipolarophilic Activity of Aromatic Bonds towards an Azomethine Ylide. Tetrahedron Letters, 12, 481-484. https://doi.org/10.1016/S0040-4039(01)96474-3
- 24. Lee, S., Chataigner, I. and Piettre, S.R. (2011) Facile Dearomatization of Nitrobenzene Derivatives and Other Nitroarenes with N-Benzyl Azomethine Ylide. Angewandte Chemie International Edition, 50, 472-476. https://doi.org/10.1002/anie.201005779
- 25. Lee, S., Diab, S., Queval, P., Sebban, M., Chataigner, I. and Piettre, S.R. (2013) Aromatic C=C Bonds as Dipolarophiles: Facile Reactions of Uncomplexed Electron-Deficient Benzene Derivatives and Other Aromatic Rings with a Non-Stabilized Azomethine Ylide. Chemistry—A European Journal, 19, 7181-7192. https://doi.org/10.1002/chem.201201238.