Advances in Material Chemistry
Vol.
07
No.
03
(
2019
), Article ID:
30026
,
9
pages
10.12677/AMC.2019.73004
Research Progress on HDAC6 Structure and Its Crystal Complexes
Aixin Geng, Hao Cui, Liyuan Zhang, Yong Zhu, Tao Lu*
China Pharmaceutical University, Nanjing Jiangsu
Received: Apr. 4th, 2019; accepted: Apr. 22nd, 2019; published: Apr. 29th, 2019

ABSTRACT
This paper mainly reviews the structure of HDAC6 and the structure of HDAC6-inhibitor crystal complexes. HDAC6 is mainly composed of NES, DD1, DD2, SE14, and ZNF-UBP, and the binding modes of inhibitors and HDAC6 protein are monodentate or double-dentate.
Keywords:HDAC6, Structure, Crystal Complex
HDAC6结构及其晶体复合物研究进展
耿爱新,崔昊,张立园,朱 雍,陆涛*
中国药科大学,江苏 南京
收稿日期:2019年4月4日;录用日期:2019年4月22日;发布日期:2019年4月29日

摘 要
本文主要综述了HDAC6的结构和HDAC6-抑制剂晶体复合物的结构。HDAC6主要由NES、DD1、DD2、SE14、ZNF-UBP几部分组成,而抑制剂和HDAC6蛋白的结合模式有单齿和双齿两种。
关键词 :HDAC6,结构,晶体复合物

Copyright © 2019 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 引言
组蛋白去乙酰化酶(HDACs)在表观遗传调控方面起重要作用,可调节组蛋白、转录因子、分子伴侣和信号分子等多种蛋白质的乙酰化水平 [1]。HDAC通常与染色质的浓缩以及多种转录基因的沉默相关 [2]。因此,HDAC抑制剂(HDACi)可用于治疗癌症,炎症 [3],神经退行性疾病 [4] 和代谢紊乱等多种疾病 [5]。目前已有五个HDAC抑制剂批准上市,SAHA、belinostat (PXD-101)、panobinostat (LBH-589)、romidepsin (FK-228)和西达本胺(Chidamide),其结构见图1。SAHA和PXD101分别于2006年和2014年被FDA批准用于治疗皮肤T细胞淋巴瘤和外周T细胞淋巴瘤;LBH589在2015年被FDA批准用于治疗多发性骨髓瘤以及皮肤和外周T细胞淋巴瘤;西达本胺在2014年被NMPA批准用于治疗复发及难治性外周T细胞淋巴瘤 [6] [7] [8] [9] [10]。
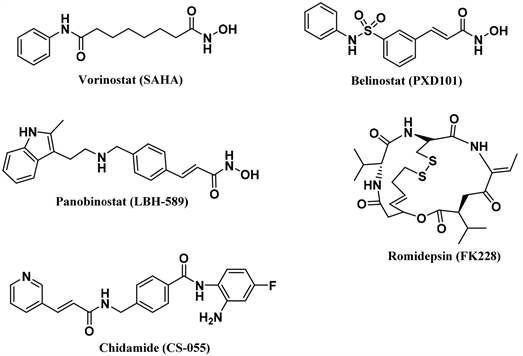
Figure 1. Structure of the HDACi approved for marketing
图1. 批准上市的HDACi的结构
在人类中已发现四类HDAC同工酶:I类包括HDAC1、2、3和8;IIa类包括HDAC4、5和7,IIb类包括HDACs 6和10;III类HDAC,命名为sirtuins 1-7;IV类只包含HDAC11 [11]。I类,II类和IV类HDAC属于精氨酸酶–去乙酰酶折叠 [12] Zn2+依赖性酶,而III类HDAC为不相关折叠NAD+依赖性酶 [13]。
上述已上市的HDACi为非选择性或部分选择性的抑制剂,它们通常会引起许多副作用,如疲劳、味觉障碍、恶心、呕吐、腹泻、骨髓抑制、心脏毒性等。这些毒性作用可能与抑制剂作用于多个种型或缺乏对同种型成员的选择性有关系,因此,越来越多的研究集中在开发选择性HDAC抑制剂作为抗癌药物。HDAC6选择性抑制剂可通过避免多个组蛋白乙酰化诱导的全基因表达变化而显示出较低毒性 [14]。因此以HDAC6为靶标的抗肿瘤药物研究已成为目前研究项目最多、应用最广的方向之一 [15] [16] [17]。本文对HDAC6的结构及其晶体复合物进行了综述,将为更好地开发选择性HDAC6抑制剂提供理论依据。
2. HDAC6的结构
HDAC6的基因位于Xp11.23中,含有1216个氨基酸,是人类HDAC家族中最大的成员。HDAC6主要由强核输出信号(NES)、两个催化结构域(CD1和CD2)、八个连续重复的十四肽(SE14)和泛素结合锌指结构域(ZnF-UBP)组成,其结构见图2。HDAC6是组蛋白去乙酰化酶家族中唯一的一个含有串联催化结构域成员。大多数HDAC位于细胞核,但由于HDAC6结构中含有NES和确保蛋白质在细胞质中的稳定锚定的SE14,所以HDAC6主要存在于细胞质中 [18],并影响细胞质中微管蛋白、cortactin [19] 和Hsp90 [20] 等非组蛋白底物的功能。抑制HDAC6会导致α-微管蛋白亚基中的K40超乙酰化进而使微管动力减弱,而HDAC6的过表达则会减少α-微管蛋白乙酰化并增加细胞运动性 [21] [22]。通过HDAC6的ZnF-UBP结构域,多聚泛素化错误折叠的蛋白质被招募到动力蛋白上再运输到聚集体。在心脏、肝脏、肾脏、睾丸、脑和胰腺中都可以观察到HDAC6的表达。

Figure 2. Structure of HDAC6
图2. HDAC6的结构
在HDAC6的两个催化结构域中,编码CD1序列中不同的点突变不会导致对α-微管蛋白去乙酰化的活性受损。但编码CD2序列中的几个点突变(W459A,D460A双取代突变,或N530A,N530D或S531A的单取代突变)会对α-微管蛋白去乙酰化活性造成严重的损害。说明CD2是影响α-微管蛋白去乙酰化活性的关键部位。此外,CD2还表现出广泛的底物特异性,而CD1则对C-末端具有乙酰基赖氨酸底物的水解具有高度特异性。但CD1、CD2水解底物都需要完整的串联结构域,单独CD1不具有催化活性,单独CD2的活性也会降低,而将单独CD1和CD2混合,也不能使两结构域的活性提高 [23]。
在CD1-CD2的晶体结构中(见图3) [24],CD1和CD2均采用经典的精氨酸酶–去乙酰酶折叠并具有保守的去乙酰酶活性位点 [12]。但CD2活性位点和底物识别区域比CD1的位点更加保守,可能是因为两个结构域具有不同的功能。这两个结构域彼此紧密相连,连接面由CD1的H13、H14、H15和H18螺旋以及CD2的H32、H33和H34螺旋,通过环连接的CD1的螺旋H17、H18和CD2的H36、H37,以及连接两个结构域的连接子(418-442)和CD2的C-末端部分(794-806)所组成。连接面的面积大约为2100 Å2。CD1和CD2在结构上非常相似,并且参与域间界面形成的结构元素也基本相同,最终形成垂直于两侧α-螺旋界面的伪双重轴。界面只有几个保守残基片段。
在单独结构中,CD1和CD2的主链也非常相似。HDAC6催化结构域N-末端和C-末端部分与其他HDAC亚型有显著区别:在每个活性位点附近催化结构域中都存在一个独特定位的10个氨基酸残基形成的α-螺旋,在CD1中为H6,CD2中为H25;以及存在于N末端部分独特的环,在CD1中是含有W78和D79的H1-H2,在CD2中是含有W459和D460的H20-H21。当用HDAC8的H6-H7取代CD2中H25时,其催化α-微管蛋白去乙酰化的活性严重降低,但HDAC6的催化功能没有受到损害,而是其选择α-微管蛋白作为底物的能力受损。当CD1中H6被取代时则对活性几乎没有影响。由于螺旋H25和后续环
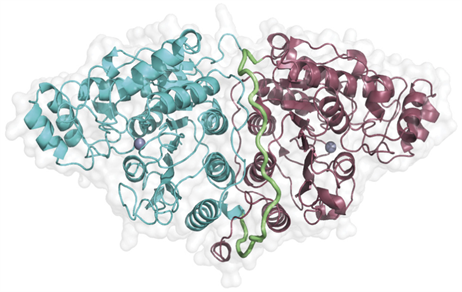
Figure 3. Crystal structure of CD1-CD2 [24]
图3. CD1-CD2的晶体结构 [24]
结构的存在,只有HDAC6存在一个开放的约14 Å宽的凹槽。HDAC6 CD1和CD2活性位点都是典型疏水空腔,空腔长度约10 Å,Zn2+离子位于空腔的底部。形成疏水空腔的残基在CD1中是P83、G201、F202和W261,在CD2中是P464、G582、F583、F643和L712。Zn2+在CD1中与D230,H232和D323配位,在CD2中与D612、H614和D705配位。此外,Zn2+还与一个水分子结合,该水分子还可与H573和H574形成氢键。配体除与Zn2+上空的配位点结合外还可与Y745相互作用。位于催化锌离子旁的酪氨酸残基可以稳定反应中间体,在CD1是Y363,在CD2中是Y745 [24]。
研究发现,人HDAC6的CD2 (hCD2)和斑马鱼HDAC6的CD2 (zCD2)活性位点区域高度相似,且活性位点氨基酸残基高度保守,只有位于活性位点外缘并与结合的配体相距很远的两个氨基酸残基不同。在zCD2与衍生自α-微管蛋白K40乙酰化位点和衍生自组蛋白H4底物的共晶结构中可以观察到催化反应中所有关键步骤(见图4)。酶-底物复合物晶体结构显示(分辨率为1.82 Å):首先,乙酰赖氨酸的可裂变羰基与Zn2+上空位点配位,且不会取代Zn2+结合的水分子,从而形成五配位的金属离子,可用于催化。此外,配体也与Y745相互作用。然后,乙酰赖氨酸的可裂变羰基经H573和Zn2+激活的水分子亲核进攻,产生一个四面体中间体。羰基氧原子通过与Zn2+的配位和与Y745形成氢键而稳定;羟基氧原子稍微远离Zn2+,但羟基氢可以与H573形成氢键作用。最后,四面体中间体裂解产生赖氨酸和乙酸产物。在这个复合物中,HDAC6 zCD2活性位点中的串联组氨酸可以作为单独的碱和酸发挥作用:H573可以辅助Zn2+激活亲核水分子,H574使离去的氨基质子化,促进底物的崩解 [23]。
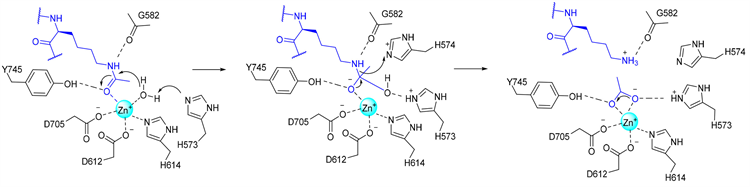
Figure 4. Key steps in the HDAC6 catalytic reaction
图4. HDAC6催化反应中关键步骤
3. HDAC6-抑制剂晶体复合物的结构
为确定选择性HDAC6抑制剂的分子特征,已有多种蛋白–抑制剂的晶体复合物结构被报道 [25] [26] [27]。总结众多HDAC6抑制剂与HDAC6 CD2结构域的晶体复合物,发现存在两种结合模式(见图5):① 以HDAC6与Ricolinostat (ACY-1215)的复合物晶体结构为代表的双齿异羟肟酸-Zn2+结合模式。② 以HDAC6与HPB、ACY-1083 [25] 、HPOB [23] 等形成复合物的晶体结构为代表的单齿异羟肟酸-Zn2+结合模式。而泛抑制剂Trichostatin A (TSA)的两个异构体分别以上述两种结合模式与CD2结构域中的Zn2+离子结合。
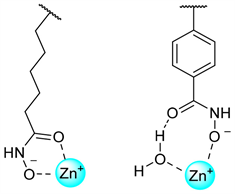
Figure 5. Schematic diagram of two binding modes of inhibitor
图5. 抑制剂的两种结合模式示意图
Ricolinostat是第一个进入临床试验的选择性HDAC6抑制剂,它与来那度胺联合用于复发和难治性多发性骨髓瘤治疗,正进行Ⅱ期临床评估 [28],而用于复发性或难治性淋巴系统恶性肿瘤的治疗,也已进入Ⅱ期临床研究阶段。HDAC6-Ricolinostat复合物的晶体结构(分辨率为1.70 Å)显示与抑制剂结合后不会使蛋白发生明显的构象变化。Ricolinostat的异羟肟酸部分的羟基氧和羰基氧以双齿模式与Zn2+配位(见图6) [25],形成规范的五元螯合物,其中Zn2+与异羟肟酸的羟基氧和羰基氧的距离分别为2.0 Å和2.4 Å。Y745的侧链作为氢键供体与异羟肟酸的羰基形成氢键,H573作为氢键供体与异羟肟酸的羟基形成氢键,此外,H574与异羟肟酸的氨基基团也可形成氢键。与Ricolinostat通过氢键作用的串联组氨酸残基中的H573处于带正电荷的咪唑鎓状态,而H574处于中性咪唑状态。Ricolinostat以两种单体存在于该酶-抑制剂复合物的不对称单元中。在这两种单体中,抑制剂Cap基团采用两种相互排斥的构象。一种构象中,抑制剂Cap基团的酰胺羰基与L1环上S531形成水介导的氢键。氨基嘧啶环上的氮原子还与D460形成水介导的氢键。在另一种构象中,抑制剂的Cap基团的酰胺羰基和H614 (Zn2+配体之一)之间形成水介导的氢键。氨基嘧啶环上的氮原子与P711的羰基形成水介导的氢键。
HDAC6-ACY-1083复合物的异羟肟酸部分则采用单齿Zn2+配位模式(见图7(a)) [25]:仅异羟肟酸的羟基氧与Zn2+ (距离为1.9 Å)配位,该羟基同时还与Y745相互作用。异羟肟酸羰基可与Zn2+结合的水分子形成氢键,该水分子还与H573和H574形成氢键。异羟肟酸氨基基团与Y745的侧链相互作用。氨基嘧啶Linker的芳环夹在F583和F643之间(见图7(b)) [25],形成π-π堆叠的相互作用。L2环上S531的羟基侧链可与抑制剂连接基团(linker)的氨基产生氢键作用。Cap基团中的二氟环己基采用椅式构象,氟原子包裹在F643侧链的边缘。抑制剂Cap区的苯基与P464和F583侧链通过范德华力相互作用。
泛HDAC抑制剂TSA的两个异构体分别以两种结合模式与Zn2+离子作用(见图8(a)) [25]。在TSA的主要构象异构体(70%占据率)中观察到二齿异羟肟酸-Zn2+配位模式(见图8(b)) [25]。Zn2+与异羟肟酸羟基氧和羰基氧配位,距离分别为2.2 Å和2.0 Å。此外,Y745与异羟肟酸的羰基形成氢键,而H573和H574
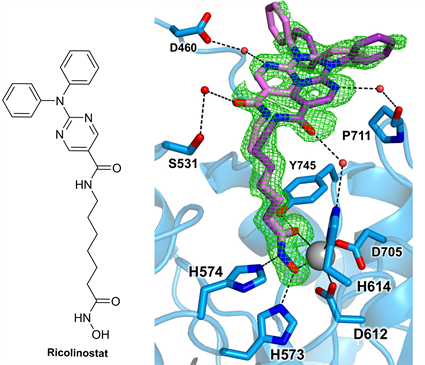
Figure 6. Ricolinostat and combination pattern of HDAC6-Ricolinostat [25]
图6. Ricolinostat及HDAC6-Ricolinostat复合物结合模式图 [25]
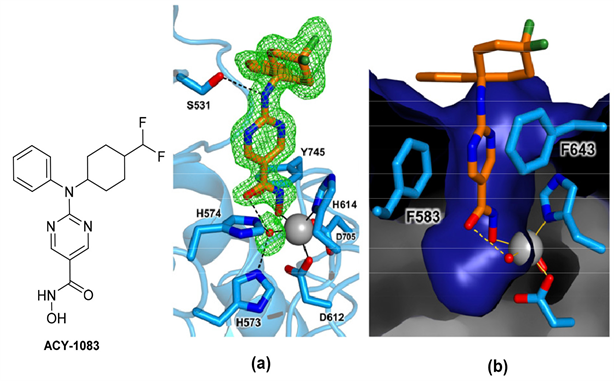
Figure 7. ACY-1083 and combination pattern of HDAC6-ACY-1083 [25]
图7. ACY-1083及HDAC6-ACY-1083复合物结合模式图 [25]
与异羟肟酸得羟基形成氢键。TSA的次要构象异构体(30%占据率)采用与HDAC6选择性抑制剂如HPOB,HPB和ACY-1083类似的单齿异羟肟酸-Zn2+配位模式(见图8(c)) [25]。TSA的异羟肟酸基团仅通过其羟基与Zn2+配位。与Zn2+结合的水分子与异羟肟酸的羰基形成氢键。Y745的羟基与异羟肟酸的亚氨基和羟基也能形成氢键,距离分别为2.7 Å和2.8 Å。除了异羟肟酸基团的构象和取向差异外,TSA的主要和次要构象没有其他明显区别。二甲基庚二烯的Linker和对二甲基氨基苯基的Cap以相同的方式结合在两种构象中。而且,单齿异羟肟酸-Zn2+结合模式的能量仅比二齿异羟肟酸酯-Zn2+结合模式高0.5 kcal/mol。
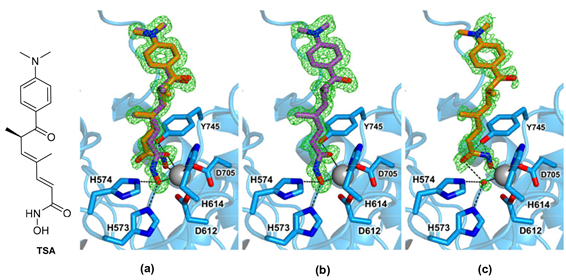
Figure 8. TSA and combination pattern of HDAC6-TSA [25]
图8. TSA及HDAC6-TSA复合物结合模式图 [25]
4. 小结
对HDAC6的结构以及HDAC6-配体的晶体复合物的解析和研究将有利于进一步开发活性和选择性更优的小分子抑制剂。与pan-HDAC抑制剂TSA相比,对HDAC6的选择性可能是由庞大的Cap或连接基团决定。HDAC6选择性抑制剂的linker区基团可与S531形成直接或水介导的氢键。该残基接受来自乙酰基-L-赖氨酸底物的主链氨基基团的氢键,并且这种相互作用对于HDAC6活性位点是独特的。因此,靶向作用于这些特异性位点的抑制剂可表现出与HDAC6结合的高选择性和强活性。抑制剂Cap基团与HDAC6活性位点口袋的相互作用揭示了L1环中的结构差异也有助于提高对HDAC6的选择性。因此,设计合适的Linker和Cap基团,特异性地作用于这两个位点将有助于提高对HDAC6作用的选择性。
文章引用
耿爱新,崔 昊,张立园,朱 雍,陆 涛. HDAC6结构及其晶体复合物研究进展
Research Progress on HDAC6 Structure and Its Crystal Complexes[J]. 材料化学前沿, 2019, 07(03): 29-37. https://doi.org/10.12677/AMC.2019.73004
参考文献
- 1. Sun, J. (2003) Measurement of Histone Acetyltransferase and Histone Deacetylase Activities and Kinetics of Histone Acetylation. Methods, 31, 12-23.
https://doi.org/10.1016/S1046-2023(03)00083-5 - 2. Xu, Z., Li, H. and Jin, P. (2012) Epigenetics-Based Therapeutics for Neurodegenerative Disorders. Current Translational Geriatrics and Experimental Gerontology Reports, 1, 229-236.
https://doi.org/10.1007/s13670-012-0027-0 - 3. Shakespear, M.R., Halili, M.A., Irvine, K.M., Fairlie, D.P. and Sweet, M.J. (2011) Histone Deacetylases as Regulators of Inflammation and Immunity. Trends in Immunology, 32, 335-343.
https://doi.org/10.1016/j.it.2011.04.001 - 4. Chuang, D.M., Leng, Y., Marinova, Z., Kim, H.J. and Chiu, C.T. (2009) Multiple Roles of HDAC Inhibition in Neurodegenerative Conditions. Trends in Neurosciences, 32, 591-601.
https://doi.org/10.1016/j.tins.2009.06.002 - 5. Bhaumik, S.R., Smith, E. and Shilatifard, A. (2007) Covalent Modifications of Histones during Development and Disease Pathogenesis. Nature Structural & Molecular Biology, 14, 1008-1016.
https://doi.org/10.1038/nsmb1337 - 6. Dong, M., Ning, Z.Q., Xing, P.Y., Xu, J.L., Cao, H.X., Dou, G.F., Meng, Z.Y., Shi, Y.K., Lu, X.P. and Feng, F.Y. (2012) Phase I Study of Chidamide (CS055/HBI-8000), a New Histone Deacetylase Inhibitor, in Pa-tients with Advanced Solid Tumors and Lymphomas. Cancer Chemotherapy and Pharmacology, 69, 1413-1422.
https://doi.org/10.1007/s00280-012-1847-5 - 7. Garnock-Jones, K.P. (2015) Panobinostat: First Global Approval. Drugs, 75, 695-704.
https://doi.org/10.1007/s40265-015-0388-8 - 8. Marks, P.A. and Breslow, R. (2007) Dimethyl Sulfoxide to Vorinostat: De-velopment of This Histone Deacetylase Inhibitor as an Anticancer Drug. Nature Biotechnology, 25, 84-90.
https://doi.org/10.1038/nbt1272 - 9. Shi, Y., Dong, M., Hong, X., Zhang, W., Feng, J., Zhu, J., Yu, L., Ke, X., Huang, H., Shen, Z., Fan, Y., Li, W., Zhao, X., Qi, J., Huang, H., Zhou, D., Ning, Z. and Lu, X. (2015) Results from a Multicenter, Open-Label, Pivotal Phase II Study of Chidamide in Relapsed or Refractory Peripheral T-Cell Lymphoma. Annals of Oncology, 26, 1766-1771.
https://doi.org/10.1093/annonc/mdv237 - 10. VanderMolen, K.M., McCulloch, W., Pearce, C.J. and Oberlies, N.H. (2011) Romidepsin (Istodax, NSC 630176, FR901228, FK228, Depsipeptide): A Natural Product Recently Approved for Cutaneous T-Cell Lymphoma. The Journal of Antibiotics (Tokyo), 64, 525-531.
https://doi.org/10.1038/ja.2011.35 - 11. Gregoretti, I.V., Lee, Y.M. and Goodson, H.V. (2004) Molecular Evolution of the Histone Deacetylase Family: Functional Implications of Phylogenetic Analysis. Journal of Molecular Biology, 338, 17-31.
https://doi.org/10.1016/j.jmb.2004.02.006 - 12. Lombardi, P.M., Cole, K.E., Dowling, D.P. and Christianson, D.W. (2011) Structure, Mechanism, and Inhibition of Histone Deacetylases and Related Metalloenzymes. Current Opinion in Structural Biology, 21, 735-743.
https://doi.org/10.1016/j.sbi.2011.08.004 - 13. Yuan, H. and Marmorstein, R. (2012) Structural Basis for Sirtuin Activity and Inhibition. The Journal of Biological Chemistry, 287, 42428-42435.
https://doi.org/10.1074/jbc.R112.372300 - 14. Zeb, A., Park, C., Rampogu, S., Son, M., Lee, G. and Lee, K.W. (2019) Structure-Based Drug Designing Recommends HDAC6 Inhibitors to Attenuate Microtubule-Associated Tau-Pathogenesis. ACS Chemical Neuroscience, 10, 1326-1335.
https://doi.org/10.1021/acschemneuro.8b00405 - 15. Lee, H.Y., Nepali, K., Huang, F.I., Chang, C.Y., Lai, M.J., Li, Y.H., Huang, H.L., Yang, C.R. and Liou, J.P. (2018) (N-Hydroxycarbonylbenylamino)quinolines as Selective Histone Deacetylase 6 Inhibitors Suppress Growth of Multiple Myeloma in Vitro and in Vivo. Journal of Medicinal Chemistry, 61, 905-917.
https://doi.org/10.1021/acs.jmedchem.7b01404 - 16. Lee, H.Y., Fan, S.J., Huang, F.I., Chao, H.Y., Hsu, K.C., Lin, T.E., Yeh, T.K., Lai, M.J., Li, Y.H., Huang, H.L., Yang, C.R. and Liou, J.P. (2018) 5-Aroylindoles Act as Selective Histone Deacetylase 6 In-hibitors Ameliorating Alzheimer’s Disease Phenotypes. Journal of Medicinal Chemistry, 61, 7087-7102.
https://doi.org/10.1021/acs.jmedchem.8b00151 - 17. Vogerl, K., Ong, N., Senger, J., Herp, D., Schmidtkunz, K., Marek, M., Muller, M., Bartel, K., Shaik, T.B., Porter, N.J., Robaa, D., Christianson, D.W., Romier, C., Sippl, W., Jung, M. and Bracher, F. (2019) Synthesis and Biological Investigation of Phenothiazine-Based Benzhydroxamic Acids as Selective Histone Deacetylase 6 Inhibitors. Journal of Medicinal Chemistry, 62, 1138-1166.
https://doi.org/10.1021/acs.jmedchem.8b01090 - 18. Boyault, C., Sadoul, K., Pabion, M. and Khochbin, S. (2007) HDAC6, at the Crossroads between Cytoskeleton and Cell Signaling by Acetylation and Ubiquitination. Oncogene, 26, 5468-5476.
https://doi.org/10.1038/sj.onc.1210614 - 19. Zhang, X., Yuan, Z., Zhang, Y., Yong, S., Salas-Burgos, A., Koomen, J., Olashaw, N., Parsons, J.T., Yang, X.J., Dent, S.R., Yao, T.P., Lane, W.S. and Seto, E. (2007) HDAC6 Modulates Cell Motility by Altering the Acetylation Level of Cortactin. Molecular Cell, 27, 197-213.
https://doi.org/10.1016/j.molcel.2007.05.033 - 20. Kovacs, J.J., Murphy, P.J., Gaillard, S., Zhao, X., Wu, J.T., Nicchitta, C.V., Yoshida, M., Toft, D.O., Pratt, W.B. and Yao, T.P. (2005) HDAC6 Regulates Hsp90 Acetylation and Chaperone-Dependent Activa-tion of Glucocorticoid Receptor. Molecular Cell, 18, 601-607.
https://doi.org/10.1016/j.molcel.2005.04.021 - 21. Szyk, A., Deaconescu, A.M., Spector, J., Goodman, B., Valenstein, M.L., Ziolkowska, N.E., Kormendi, V., Grigorieff, N. and Roll-Mecak, A. (2014) Molecular Basis for Age-Dependent Microtubule Acetylation by Tubulin Acetyltransferase. Cell, 157, 1405-1415.
https://doi.org/10.1016/j.cell.2014.03.061 - 22. Woan, K.V., Lienlaf, M., Perez-Villaroel, P., Lee, C., Cheng, F., Knox, T., Woods, D.M., Barrios, K., Powers, J., Sahakian, E., Wang, H.W., Canales, J., Marante, D., Smalley, K.S.M., Bergman, J., Seto, E., Kozikowski, A., Pinilla-Ibarz, J., Sarnaik, A., Celis, E., Weber, J., Sotomayor, E.M. and Villagra, A. (2015) Targeting Histone Deacetylase 6 Mediates a Dual Anti-Melanoma Effect: Enhanced Antitumor Immunity and Impaired Cell Proliferation. Molecular Oncology, 9, 1447-1457.
https://doi.org/10.1016/j.molonc.2015.04.002 - 23. Hai, Y. and Christianson, D.W. (2016) Histone Deacetylase 6 Structure and Molecular Basis of Catalysis and Inhibition. Nature Chemical Biology, 12, 741-747.
https://doi.org/10.1038/nchembio.2134 - 24. Miyake, Y., Keusch, J.J., Wang, L., Saito, M., Hess, D., Wang, X., Melancon, B.J., Helquist, P., Gut, H. and Matthias, P. (2016) Structural Insights into HDAC6 Tubulin Deacetylation and Its Selective Inhibition. Nature Chemical Biology, 12, 748-754.
https://doi.org/10.1038/nchembio.2140 - 25. Porter, N.J., Mahendran, A., Breslow, R. and Christianson, D.W. (2017) Unusual Zinc-Binding Mode of HDAC6-Selective Hydroxamate Inhibitors. Proceedings of the National Academy of Sciences of the United States of America, 114, 13459-13464.
https://doi.org/10.1073/pnas.1718823114 - 26. Ferreira de Freitas, R., Harding, R.J., Franzoni, I., Ravichandran, M., Mann, M.K., Ouyang, H., Lautens, M., Santhakumar, V., Arrowsmith, C.H. and Schapira, M. (2018) Identification and Structure-Activity Relationship of HDAC6 Zinc-Finger Ubiquitin Binding Domain Inhibitors. Journal of Medicinal Chemistry, 61, 4517-4527.
https://doi.org/10.1021/acs.jmedchem.8b00258 - 27. Harding, R.J., Ferreira de Freitas, R., Collins, P., Franzoni, I., Ravichandran, M., Ouyang, H., Juarez-Ornelas, K.A., Lautens, M., Schapira, M., von Delft, F., Santhakumar, V. and Arrowsmith, C.H. (2017) Small Molecule Antagonists of the Interaction between the Histone Deacetylase 6 Zinc-Finger Domain and Ubiquitin. Journal of Medicinal Chemistry, 60, 9090-9096.
https://doi.org/10.1021/acs.jmedchem.7b00933 - 28. Yoo, J., Kim, S.J., Son, D., Seo, H., Baek, S.Y., Maeng, C.Y., Lee, C., Kim, I.S., Jung, Y.H., Lee, S.M. and Park, H.J. (2016) Computer-Aided Identification of New Histone Deacetylase 6 Selective Inhibitor with Anti-Sepsis Activity. European Journal of Medicinal Chemistry, 116, 126-135.
https://doi.org/10.1016/j.ejmech.2016.03.046
NOTES
*通讯作者。