Advances in Material Chemistry
Vol.
09
No.
03
(
2021
), Article ID:
43635
,
9
pages
10.12677/AMC.2021.93009
二维多孔材料C2N理论研究综述
孔程程*,黄华荃
浙江师范大学含氟新材料研究所,浙江 金华
收稿日期:2021年6月3日;录用日期:2021年6月16日;发布日期:2021年7月1日

摘要
C2N的结构与石墨烯相似,主要由sp2杂化的碳原子组成。但与石墨烯不同的是,C2N是一种平面存在周期性小孔,孔的边界由氮原子组成,导致电子在氮原子上富集的材料。由于C2N具有表面积高、结晶度良好和离子传输快速等优点,在催化、纳米电子传感器、气体存储和电池等领域有着广泛的应用。C2N材料是当前的一个研究热点之一,负载的过渡金属或贵金属元素作为可能的催化活性位点受到了广泛的关注。最近几年在合成和应用C2N材料方面已经取得了很大进展,然而对材料中金属纳米颗粒的几何结构、电子性质及其形成机理仍不清楚,此外对催化反应的微观机理缺乏深入的认识。本文综述了C2N体系的几何形状和稳定性,电子结构分析,选择合适的理论计算方法可探究在C2N上锚定金属原子体系的催化性能,为我们提供了结构和性能等方面的重要信息,从而为设计出性能更好的催化剂提供借鉴与指导意义。
关键词
C2N,几何结构,电子结构,甲酸,理论化学

A Summary of C2N Theoretical Research on Two-Dimensional Porous Materials
Chengcheng Kong*, Huaquan Huang
Institute of Advanced Fluorine-Containing Materials, Zhejiang Normal University, Jinhua Zhejiang

Received: Jun. 3rd, 2021; accepted: Jun. 16th, 2021; published: Jul. 1st, 2021

ABSTRACT
The structure of C2N is similar to graphene, mainly composed of sp2 hybridized carbon atoms. But unlike graphene, there are periodic pores in the C2N plane, and the boundary of the pores is composed of nitrogen atoms, resulting in a material where electrons are enriched in nitrogen atoms. Because C2N has the advantages of high surface area, good crystallinity and fast ion transmission, it has a wide range of energy and environmental applications in the fields of catalysis, nanoelectronic sensors, gas storage, and batteries. C2N materials are currently a research hotspot, and supported transition metals or metal-free elements have received extensive attention as possible catalytically active sites. In recent years, great progress has been made in the synthesis and application of C2N materials. However, the geometric structure, electronic properties and formation mechanism of metal nanoparticles in the materials are still unclear. In addition, there is a lack of in-depth understanding of the microscopic mechanism of catalytic reactions. This article reviews the geometry and stability of the C2N system, analysis of the electronic structure, and selects appropriate theoretical calculation methods to explore the catalytic performance of the metal atom system anchored to C2N. It provides us with important information on the structure and performance. It provides reference and guidance for the design of better performance catalysts.
Keywords:C2N, Geometric Structure, Electronic Structure, Formic Acid, Theoretical Chemistry

Copyright © 2021 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/


1. 引言
载体的电子性质和结构性能对金属原子的分散性、稳定性和吸附扩散等有着重要的影响。一般来说,理想的载体能够很好地包裹和分散金属纳米粒子,促进反应物的吸附,加速产物的扩散。具有独特几何和电子结构的二维(2D)材料已成为人们的关注焦点 [1]。近年来,C2N材料受到了广泛的关注,该类催化剂具有大比表面积,将单个原子锚定在表面上而创建了更多的活性位点;有助于反应分子从单原子的两侧吸附和扩散,因此它已被广泛应用于各种重要化学反应,例如一些重要的电池反应以及HER,ORR和NRR等反应中 [2]。我们知道原始石墨烯和BN作为金属原子的底物已被广泛研究 [3],并且我们发现金属原子在这些载体表面很容易扩散和凝聚,通过研究我们发现引入空位和孔可以解决该问题,但是在大规模应用中精确雕刻空位和孔非常困难。2015年,一种新颖的2D分层材料,这种材料是一个分层的二维网络结构 [4],由于具有天然的孔结构,C2N-h2D可以充当金属簇甚至单原子催化剂的载体。
最近,合成了分散在C2N-h2D的孔中的Ru纳米颗粒的复合物(Ru@C2N),对HER具有优异的催化活性 [5]。此外,C2N具有类似于石墨烯的结构,其中所有碳原子的1/3被吡嗪氮原子取代。这导致C2N具有共价的“沸石样”结构,其中的孔是规则的,理想情况下,有12个原子。通过桥接氮原子连接苯环的C2N独特的多孔结构在苯环中产生了一个大的π电子,并在N原子上产生了固有的电子密度。将N原子引入碳晶格可增强碳材料的性能,例如化学和热稳定性,能带位置,催化效率和氧化稳定性 [6] [7] [8],并有助于与液体,溶剂化离子或气体的特定相互作用 [9] [10]。C2N可以使用常规方法制备,包括溶剂热,离子热和直接冷凝。但是,其孔隙率,边缘和结构的规则性可能会发生变化。低温下的溶剂热法促进了规则的2D晶体结构的形成,这有望将带隙扩大到半导体应用的理想水平 [11] [12]。
Baek及其同事在使用C2N单层作为催化剂载体方面取得了最新进展,他们报告说,分离出的Ru纳米颗粒可以分散在C2N的氮化孔中,对C2N具有良好的催化活性 [13]。同时,在实验中也有人们合成出了Fe@C2N,其表现出优异的电催化氧还原活性 [14]。从理论上讲,一些研究还报道了在C2N单层上单金属原子的沉积可作为各种反应的催化剂,例如,Li、Ma等 [15] [16],提出锚定在C2N单层上的单金属原子是有希望的析氧电催化剂,表明嵌入单层的单个Sc,Ti,V,Cr和Mn原子有望用于CO氧化,而Mo@C2N具有用于氨合成的良好催化性能。本文主要综述了研究C2N材料的吸附构型,吸附能,电荷转移和电子性质和相应的反应机理。我们希望我们的工作能够揭示基于C2N的材料的设计的一些理论指导。
2. 催化剂材料分析
2.1. 几何形状和稳定性
首先研究了TM原子锚定的C2N单层的几何形状和稳定性。对于单个TM原子锚定的C2N单层,考虑了三个可能的锚定位点,如图1所示 [1]:i) TM原子位于孔的中心(图1(a));ii) TM原子与两个N原子键合(图1(b));或iii) TM原子与三个N原子键合(图1(c))。对于半径小的TM原子(Mn,Co,Ni,Cu,Ru和Pd),最稳定的位置是TM原子与两个N原子键合的位置(位置ii)。随着半径的增加,最稳定的位点将更改为位点iii。对于半径较大的Au和Ag原子,位点i是最稳定的,也就如图1(a)所示的结构。
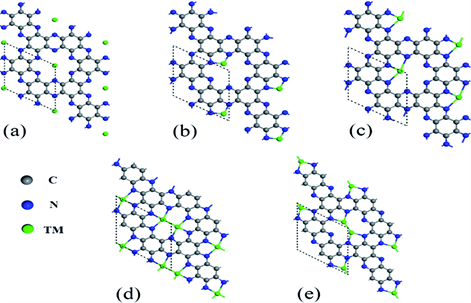
Figure 1. Three possible anchoring sites for single TM atoms: (a) at the center of the holes, (b) bonding with two N atoms, and (c) bonding with three N atoms. Two possible anchoring sites for double TM atoms: (d) and (e) [1]
图1. 单个TM原子的三个可能的锚定位点:(a)在孔的中心,(b)与两个N原子键合,以及(c)与三个N原子键合,(d)和(e)双TM原子的两个可能的锚定位点 [1]
2.2. 电子结构分析
电导率是评估电催化性能的重要指标。优异的电导率可确保有效HER和OER的良好电荷转移。因此,如图2所示 [1],计算原始C2N和TMx@C2N的能带结构以估计电导率。预测的状态密度(PDOS)显示,价带最大(VBM)和导带最小(CBM)均由杂化Cp和Np状态主导。大的带隙将不利于电催化反应的电荷转移。但是,在锚定TM原子后,几乎所有TMx@C2N单层(Ru@C2N除外)都表现出金属性能。我们通过分析金属Cu锚定在C2N载体上,我们会发现这个特点:
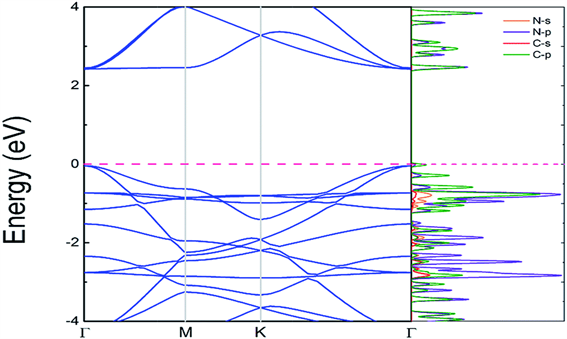
Figure 2. Structures and projected density of states (PDOS) of the C2N monolayer calculated at the HSE06 level. The Fermi level is set to zero [1]
图2. HSE06水平计算的C2N单层结构和投影态密度(PDOS),费米能级设置为零 [1]
在所有活性位点中,单一金属更倾向于通过与N(1)和N(2)原子键合嵌入C2N单层腔中,这与先前的研究一致,根据态密度(DOS)分析,如图3(b)所示 [1],与原始C2N相比,Cu@C2N的费米能级向右移动进入LUMO轨道,这时候催化剂则很容易接受电子。此外我们还分析了双金属Cu锚定在C2N上的结构;即Cu2@C2N我们计算Cu2@C2N的能带结构和部分态密度(PDOS)(图4),并特别检查了其能带间隙和费米能级附近能带中的电荷分布,我们计算的原始C2N层的带隙为1.77 eV。Cu2@C2N层不包含任何磁矩。Cu2@C2N层的带隙大大减小到0.58 eV,因此表明导电性也得到了很好的改善。此外,计算的PDOS表明在Cu的3d和N的2p轨道之间存在强杂交,进一步证明了Cu二聚体与C2N层之间的强相互作用。因此我们推测金属原子锚定在C2N这种材料上时可以进行一系列的催化反应。
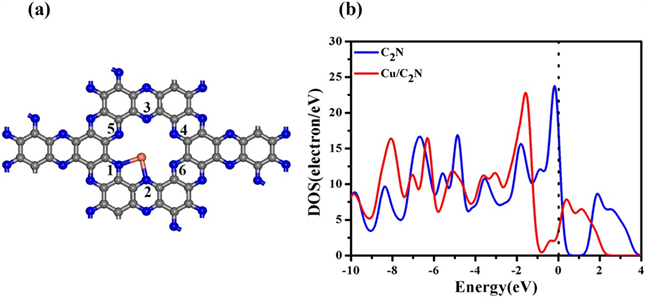
Figure 3. (a) The top view of the Cu-C2N monolayer. The gray, blue and orange spheres represent the C, N and Cu atoms, respectively. (b) The DOS of the C2N and Cu-C2N monolayers [1]
图3. (a) Cu-C2N单层的俯视图。灰色、蓝色和橙色球体分别代表C、N和Cu原子。(b) C2N和Cu-C2N单层的DOS [1]

Figure 4. (a) Optimized structure, (b) band structure, and (c) partial density of states (PDOS) of a Cu dimer supported on a porous C2N layer. The Fermi level was set at 0 [1]
图4. (a)优化的结构,(b)能带结构,和(c)支撑在多孔C2N层上的Cu二聚体的部分态密度(PDOS), 费米能级设为0 [1]
3. 化学反应机理研究
M@C2N催化HCOOH氧化反应机理
文章中首先模拟了单个TMx+离子(Co2+,Ni2+和Cu2+)在小的C2N模型簇上的沉积。这种C2N-TMx+系统的形成非常稳定,这是因为TM离子与两个键长为1.99~2.18 Å的N原子之间的牢固结合。计算出的锚定在C2N上的Co2+,Ni2+和Cu2+的结合能均大于14 eV (表1),表明存在很强的耦合。发现沿着一个空位圆从一个吸附位置到相邻一个吸附位置的扩散势垒仅为0.07 eV,这表明离子容易沿着空位圆漂移。但是,从一个孔位到相邻孔位的扩散势垒高达3.91 eV,表明TMx+与氮之间的强相互作用阻止了TMx+的聚集。

Table 1. Computed binding energies of TMx+ on C2N, adsorption energies of the HCOOH molecule on C2N-TMx+, charges extracted from C2N to TMx+ (C2N→TMx+) in the hybrid systems, and charges extracted from HCOOH to C2N-TMx+ in the adsorption configuration
表1. 计算TMx+在C2N上的结合能、HCOOH分子在C2N-TMx+上的吸附能、混合系统中从C2N提取到TMx+ (C2N→TMx+)的电荷,以及在吸附配置中从HCOOH提取到C2N–TMx+的电荷 [17]
并且在载体和金属离子之间存在着很强的电子转移情况,更进一步的表明了金属离子能够稳定的和载体键合。
然后,我们模拟了HCOOH在C2N-TMx+上最稳定的吸附状态 [17],以探索脱氢的可能反应途径。如图5(a)所示,HCOOH通过两个位点与C2N-TMx+杂化物结合:羰基O位于TMx+离子的顶部,而OH基团的H与C2N的N原子形成氢键,但未连接到TMx+。HCOOH的吸附能为0.48~1.15 eV,表明化学键很强(表1),顺序为C2N-Ni2+ > C2N-Co2+ > C2N-Cu2+。吸附后,如果与C2N-TMx+结合,则O-H键的长度将从游离HCOOH中的0.98 Å延长至1.17~1.34 Å。此外,EDD分析(图5(b))显示,在N和TMx+的两个结合位点积累了有效电荷,这意味着HCOOH分子可能通过这两个活性位点与C2N-TMx+反应。
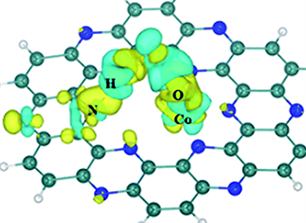
Figure 5. (a) Configuration of HCOOH adsorbed on C2N-TMx+. (b) Electron density difference for HCOOH adsorbed on C2N-Co2+ [17]
图5. (a) HCOOH吸附在C2N-TMx+上的构型。(b)吸附在C2N-Co2+上的HCOOH的电子密度差异 [17]
通过计算发现,在金属Co上进行甲酸的脱氢反应机理及能垒大小如下图6所示,我们会发现甲酸的脱氢反应的决速步骤为第二个氢原子解离的那一步,即图中的TS3-4,能垒为0.30 eV。
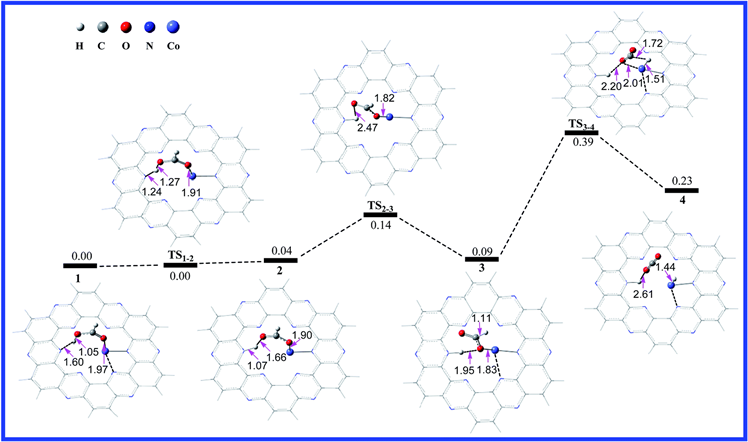
Figure 6. Calculated potential energy profile of the HCOOH dehydrogenation reaction on C2N-Co2+ with the optimized geometries of intermediates and transition states involved in the reaction. The relative free energies are given in eV. The distances are in Å [17]
图6. C2N-Co2+上HCOOH脱氢反应的计算势能分布,其中包含反应中优化的中间体和过渡态的几何形状。相对自由能以eV给出。距离以Å为单位 [17]
此外,还计算了不同的金属锚定在C2N材料上对甲酸的脱氢反应,金属Cu和金属Ni的反应,结果如下图所示:
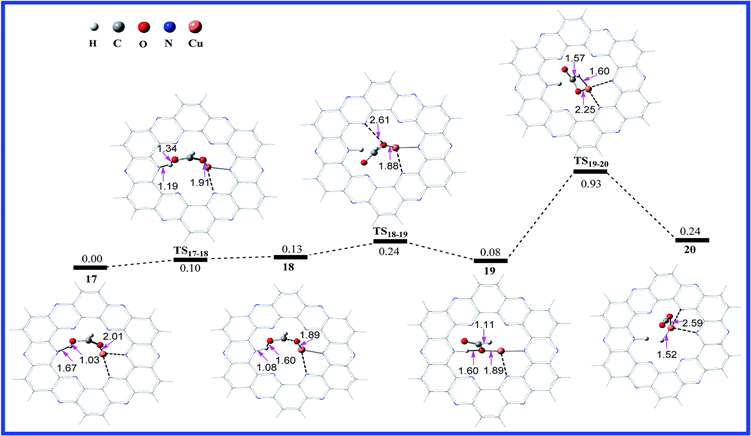
Figure 7. Calculated potential energy profile of the HCOOH dehydrogenation reaction on C2N-Cu2+ with the optimized geometries of intermediates and transition states involved in the reaction. The relative free energies are given in eV. The distances are in Å [17]
图7. C2N-Cu2+上HCOOH脱氢反应的计算势能分布,其中包括反应中优化的中间体和过渡态的几何形状。相对自由能以eV给出。距离以Å为单位 [17]
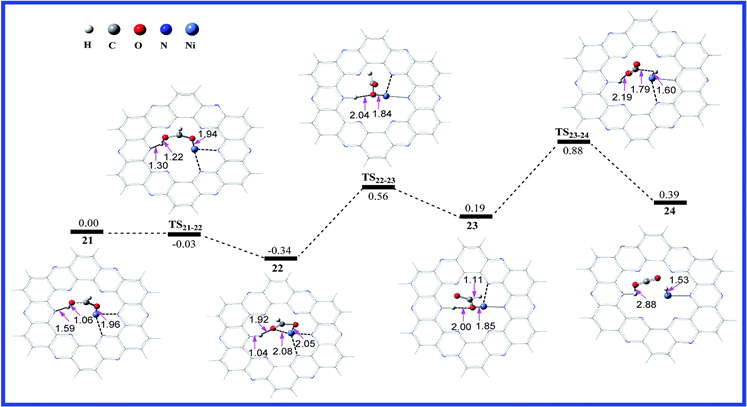
Figure 8. Calculated potential energy profile of the HCOOH dehydrogenation reaction on C2N-Ni2+ with the optimized geometries of intermediates and transition states involved in the reaction. The relative free energies are given in eV. The distances are in Å [17]
图8. 计算出的C2N-Ni2+上HCOOH脱氢反应的势能分布以及反应中涉及的中间体和过渡态的优化几何形状。相对自由能以eV给出。距离以Å为单位 [17]
我们会发现在金属Cu和Ni上的决速步能垒分别为0.85 eV和0.69 eV,因此我们会发现将金属原子锚定在C2N上来进行甲酸的脱氢反应是一种良好的催化剂。除此之外,还有一些课题组利用C2N载体来进行一些加氢反应,例如:CO2与H2反应形成甲酸的反应 [18] [19],还有一些电催化 [20] 的反应等等,都取得了很好的效果。
4. 总结和展望
本文综述了C2N体系的几何形状和稳定性,电子结构分析,选择合适的理论计算方法可探究在C2N上锚定金属原子体系的催化性能,最后对一些具体反应的反应机理的进行探讨。利用计算建模的手段可以将催化活性与反应过程中发生变化的性质等联系起来,使我们更加清晰地认识催化剂的结构与性能之间的关系,从而揭示催化活性的本质,为我们能够有效地调控和优化催化剂性能提供了理论指导和借鉴意义。
文章引用
孔程程,黄华荃. 二维多孔材料C2N理论研究综述
A Summary of C2N Theoretical Research on Two-Dimensional Porous Materials[J]. 材料化学前沿, 2021, 09(03): 71-79. https://doi.org/10.12677/AMC.2021.93009
参考文献
- 1. Ma, J., Gong, H., Zhang, T., Yu, H., Zhang, R., Liu, Z., Yang, G., Sun, H., Tang, S. and Qiu, Y. (2019) Hydrogenation of CO2 to Formic Acid on the Single Atom Catalysis Cu/C2N: A First Principles Study. Applied Surface Science, 488, 1-9.
https://doi.org/10.1016/j.apsusc.2019.03.187 - 2. Ling, C., Shi, L., Ouyang, Y., Zeng, X.C. and Wang, J. (2017) Nanosheet Supported Single-Metal Atom Bifunctional Catalyst for Overall Water Splitting. Nano Letters, 17, 5133-5139.
https://doi.org/10.1021/acs.nanolett.7b02518 - 3. Feng, B., Zhang, J., Zhong, Q., Li, W., Li, S., Li, H., Cheng, P., Meng, S., Chen, L. and Wu, K. (2016) Experimental Realization of Two-Dimensional Boron Sheets. Nature Chemistry, 8, 563-568.
https://doi.org/10.1038/nchem.2491 - 4. Mahmood, J., Lee, E.K., Jung, M., Shin, D., Jeon, I.Y., Jung, S.M., Choi, H.J., Seo, J.M., Bae, S.Y., Sohn, S.D., Park, N., Oh, J.H., Shin, H.J. and Baek, J.B. (2015) Nitrogenated Holey Two-Dimensional Structures. Nature Communications, 6, Article No. 6486.
https://doi.org/10.1038/ncomms7486 - 5. Mahmood, J., Li, F., Jung, S.M., Okyay, M.S., Ahmad, I., Kim, S.J., Park, N., Jeong, H.Y. and Baek, J.B. (2017) An Efficient and pH-Universal Ruthenium-Based Catalyst for the Hydrogen Evolution Reaction. Nature Nanotechnology, 12, 441-446.
https://doi.org/10.1038/nnano.2016.304 - 6. Tian, Z., Fechler, N., Oschatz, M., Heil, T., Schmidt, J., Yuan, S. and Antonietti, M. (2018) C2NxO1−X Framework Carbons with Defined Microporosity and Co-Doped Functional Pores. Journal of Materials Chemistry A, 6, 19013-19019.
https://doi.org/10.1039/C8TA03213K - 7. Seema, H., Kemp, K.C., Le, N.H., Park, S.-W., Chandra, V., Lee, J.W. and Kim, K.S. (2014) Highly Selective CO2 Capture by S-Doped Microporous Carbon Materials. Carbon, 66, 320-326.
https://doi.org/10.1016/j.carbon.2013.09.006 - 8. Paraknowitsch, J.P. and Thomas, A. (2013) Doping Carbons beyond Nitrogen: An Overview of Advanced Heteroatom Doped Carbons with Boron, Sulphur and Phosphorus for Energy Applications. Energy & Environmental Science, 6, 2839-2855.
https://doi.org/10.1039/c3ee41444b - 9. Guan, Z., Lian, C.S., Hu, S., Ni, S., Li, J. and Duan, W. (2017) Tunable Structural, Electronic, and Optical Properties of Layered Two-Dimensional C2N and MoS2 van der Waals Heterostructure as Photovoltaic Material. The Journal of Physical Chemistry C, 121, 3654-3660.
https://doi.org/10.1021/acs.jpcc.6b12681 - 10. Bafekry, A., Stampfl, C., Ghergherehchi, M. and Shayesteh, S.F. (2020) A First-Principles Study of the Effects of Atom impurities, Defects, Strain, Electric Field and Layer Thickness on the Electronic and Magnetic Properties of the C2N Nanosheet. Carbon, 157, 371-384.
https://doi.org/10.1016/j.carbon.2019.10.038 - 11. Mahmood, J., Li, F., Kim, C., Choi, H.J., Gwon, O., Jung, S.-M., Seo, J.-M., Cho, S.-J., Ju, Y.W., Jeong, H.Y., Kim, G. and Baek, J.-B. (2018) Fe@C2N: A Highly-Efficient Indirect-Contact Oxygen Reduction Catalyst. Nano Energy, 44, 304-310.
https://doi.org/10.1016/j.nanoen.2017.11.057 - 12. Li, X., Cui, P., Zhong, W., Li, J., Wang, X., Wang, Z. and Jiang, J. (2016) Graphitic Carbon Nitride Supported Single-Atom Catalysts for Efficient Oxygen Evolution Reaction. Chemical Communications, 52, 13233-13236.
https://doi.org/10.1039/C6CC07049C - 13. Ma, D.W., Wang, Q., Yan, X., Zhang, X., He, C., Zhou, D., Tang, Y., Lu, Z. and Yang, Z. (2016) 3d Transition Metal Embedded C2N Monolayers as Promising Single-Atom Catalysts: A First-Principles Study. Carbon, 105, 463-473.
https://doi.org/10.1016/j.carbon.2016.04.059 - 14. Wang, Z., Yu, Z. and Zhao, J. (2018) Computational Screening of a Single Transition Metal Atom Supported on the C2N Monolayer for Electrochemical Ammonia Synthesis. Physical Chemistry Chemical Physics, 20, 12835-12844.
https://doi.org/10.1039/C8CP01215F - 15. Zhang, X., Chen, A., Zhang, Z., Jiao, M. and Zhou, Z. (2018) Transition Metal Anchored C2N Monolayers as Efficient Bifunctional Electrocatalysts for Hydrogen and Oxygen Evolution Reactions. Journal of Materials Chemistry A, 6, 11446-11452.
https://doi.org/10.1039/C8TA03302A - 16. Zhang, R., Li, B. and Yang, J. (2015) Effects of Stacking Order, Layer Number and External Electric Field on Electronic Structures of Few-Layer C2N-h2D. Nanoscale, 7, 14062-14070.
https://doi.org/10.1039/C5NR03895B - 17. Zhan, Y., Shen, Y., Du, Y., et al. (2017) Promotion of Iridium Complex Catalysts for HCOOH Dehydrogenation by Trace Oxygen. Kinetics and Catalysis, 58, 499-505.
https://doi.org/10.1134/S002315841705024X - 18. Kishore, M.R.A. and Ravindran, P. (2017) Enhanced Photocatalytic Water Splitting in a C2N Monolayer by C-Site Isoelectronic Substitution. ChemPhysChem, 18, 1526-1532.
https://doi.org/10.1002/cphc.201700165 - 19. Chen, Z.H., Deng, S.B., et al. (2013) Polyethylenimine-Impregnated Resin for High CO2 Adsorption: An Efficient Adsorbent for CO2 Capture from Simulated Flue Gas and Ambient Air. ACS Applied Materials & Interfaces, 5, 6937-6945.
https://doi.org/10.1021/am400661b - 20. Ssd, A., Mdd, A., Th, B., et al. (2020) Investigating CO2 Storage Properties of C2N Monolayer Functionalized with Small Metal Clusters. Journal of CO2 Utilization, 35, 1-13.
https://doi.org/10.1016/j.jcou.2019.08.014