Studies in Synthetic Chemistry
Vol.03 No.01(2015), Article ID:14925,5
pages
10.12677/SSC.2015.31001
Catalyzed Transfer Hydrogenation of Acetophenone to Phenethyl Alcohol in Water
Jiahao Long, Quan Fang, Yimei Liu, Yuting Zhang, Hongxia Ma
College of Biological, Chemical Sciences and Engineering, Jiaxing University, Jiaxing Zhejiang
Email: jxmahx@126.com
Received: Feb. 24th, 2015; accepted: Mar. 9th, 2015; published: Mar. 10th, 2015
Copyright © 2015 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



ABSTRACT
Transfer hydrogenation of acetophenone catalyzed by water-soluble ruthenium complex, RuCl2 (TPPTS)2 [TPPTS: P(m-C6H4SO3Na)3], in the presence of surfactants, was studied. The results showed that the reaction was obviously accelerated by double long-chain cationic surfactants. This was attributed to the formation of vesicles. In this vesicle system, the separation of catalyst and product is simplified after reaction, and the catalyst could be reused for at least 10 times in high conversion.
Keywords:Double Long-Chain Surfactants, Vesicles, Catalyzed Transfer Hydrogenation
水相体系中苯乙酮催化氢转移制备苯乙醇
龙嘉豪,方权,刘益美,张雨婷,马红霞
嘉兴学院生物与化学工程学院,浙江 嘉兴
Email: jxmahx@126.com
收稿日期:2015年2月24日;录用日期:2015年3月9日;发布日期:2015年3月10日

摘 要
以水溶性RuCl2(TPPTS)2 [TPPTS: P(m-C6H4SO3Na)3]为催化剂,研究了表面活性剂存在下水–有机两相体系中苯乙酮的催化氢转移反应。结果显示双长链阳离子型表面活性剂的加入,能够显著提高目标反应的转化率,同时这一反应体系使产物与催化剂的分离过程简化,有利于催化剂的循环使用,实验证实在这样的条件下,催化剂循环使用十次活性没有明显下降。
关键词 :双长链表面活性剂,泡囊,催化氢转移反应

1. 引言
羰基化合物的转移氢化是合成用途广泛的醇类化合物的重要方法之一。这类反应以醇类、甲酸(盐)、环己烯等为氢源,反应条件温和,在实验室研究和工业生产中有着广阔的应用前景。钌、铑、铱等过渡金属配合物是这类反应中应用最为广泛的催化剂。
由于反应底物的疏水性,早期的羰基化合物催化转移氢化反应基本上都是在有机溶剂中进行的,如RuCl2(PPh3)3 [1] 和[Rh(S,S)-C6P2N2][PF6] [2] 等催化剂体系。均相催化剂表现出良好的催化活性,但其主要缺点是分离比较困难、催化剂不易回收;另一方面有机溶剂有挥发性和毒性,这在工业生产中是一个比较严重的环境问题。水没有毒性,其挥发性也远比大多数有机溶剂小,循环利用也比有机溶剂容易[3] [4] 。因此,随着绿色化学的发展,研究在水溶液中进行羰基化合物的转移氢化反应将有重要意义。
采用水溶性催化剂的水相反应体系,不仅适应绿色化学的发展方向,而且可以简化产物的分离,有利于催化剂的循环使用[5] -[8] 。但是由于反应底物的烃溶性,这一水–有机两相的反应体系中存在较大的两相间传质阻碍,使得反应速率会大大降低[9] [10] ,表面活性物质的加入可以有效地解决这一问题[11] -[15] 。本文以水–有机两相体系中苯乙酮的催化氢转移反应为模型反应,在反应体系中加入不同结构类型的表面活性剂,探究表面活性剂在水–有机两相体系中的助催化作用。
2. 实验部分
2.1. 试剂
所有试剂除特殊说明外,均为市售化学试剂品,使用前进行脱氧处理。水溶性膦配体TPPTS及水溶性催化剂RuCl2(TPPTS)2均按照文献方法合成[16] ,并采用31P NMR、IR及元素分析进行表征。
2.2. 苯乙酮的催化氢转移反应
氢转移反应在带有恒温磁力搅拌的反应瓶中进行。反应烧瓶用氩气置换,依次加入钌催化剂、TPPTS、表面活性剂和KOH水溶液,搅拌溶解后,再加入氢转移试剂和反应物异丙醇和苯乙酮。反应一定时间后,用氯仿萃取出有机相,将氯仿溶液用水洗涤并用MgSO4干燥,然后用Agilent 6890氢火焰气相色谱仪分析产物组成,色谱柱为PEG-20M (30 m × 0.25 mm),苯乙酮的转化率和选择性按面积归一化法计算。
3. 结果与讨论
3.1. 不同种类表面活性剂对反应的助催化作用
不同种类表面活性剂存在下水–有机两相体系中苯乙酮氢转移反应的实验结果列于表1。在其它反应条件相同时,体系中不添加表面活性剂时,苯乙酮转化率较低,仅为37%。阳离子表面活性剂CTAB (cetyltrimethylammonium bromide,十六烷基三甲基溴化铵)和DDAB16 (dihexadecyldimethylammonium bromide,双十六烷基二甲基溴化铵)的加入,大大提高了苯乙酮的转化率,它们分别达到了68.5%和84.2%。阴离子表面活性剂SDS(sodium dodecyl sulfate,十二烷基硫酸钠)或非离子表面活性剂Tween 80加入对氢转移反应有一定的抑制作用。
阳离子表面活性剂对反应的助催化作用可能是由于表面活性剂的浓度大于临界胶束浓度(CMC)后,形成的胶束微环境大大增加了水–有机两相间的界面面积,同时胶束的阳离子头朝向水的正电场,通过静电作用将带负电荷的钌催化剂活性物种吸附于胶束的亲水界面层,从而使催化剂浓度在此界面层中大大提高。
阳离子表面活性剂对反应的助催化作用可能是由于表面活性剂的浓度大于临界胶束浓度(CMC)后,形成的胶束微环境大大增加了水–有机两相间的界面面积,同时胶束的阳离子头朝向水的正电场,通过静电作用将带负电荷的钌催化剂活性物种吸附于胶束的亲水界面层,从而使催化剂浓度在此界面层中大大提高。
双长链阳离子表面活性剂的加入,对转化率的提高最为显著。这是由于随着长链烷基数目的增多,表面活性剂的CMC(critical micelle concentration,临界胶束浓度)有较大的下降。由图1不同浓度CTAB和DDAB16溶液表面张力的变化曲线图,可以得到CTAB的CMC为8.9 × 10−4 mol·L−1,DDAB16的CMC则是2.0 × 10−4 mol·L−1。因此,当水溶液中表面活性剂的浓度相同并且超过其CMC时,双长链表面活性剂形成胶束的数量远大于单长链表面活性剂,尤其是双长链表面活性剂在水相中形成泡囊结构的胶束(如图2)是不同于单长链表面活性剂形成的球型胶束,它包含的内水相和外水相大大扩展了两相接触界面,更有利于钌催化剂在泡囊亲水表层的富集[17] 。同时泡囊中的长链烷烃形成的疏水腔使烃溶性底物更容易在其内核中溶解,因此泡囊中底物苯乙酮的局部浓度也大大提高[18] 。这样的反应微环境可以有效减小苯乙酮从烃溶性内核到达界面层的传质能量,有益于底物与催化剂的络合反应。
尽管阴离子和非离子型表面活性剂也能形成胶束增大两相间接触界面,但是SDS的阴离子头形成的

Table 1. Effect of different surfactants on the transfer hydrogenation of acetophenone
表1. 不同表面活性剂对苯乙酮氢转移反应的影响
Reaction conditions: [Ru] = 9.7 × 10−4 mol/L; [TPPTS]: [Ru] = 3:1; [Substrate]:[Catalyst]:[KOH] = 250:1:14; [Surfactant] = 2.0 × 10−3 mol/L; 2-propanol: water = 1:1; 60˚C; 6 h.
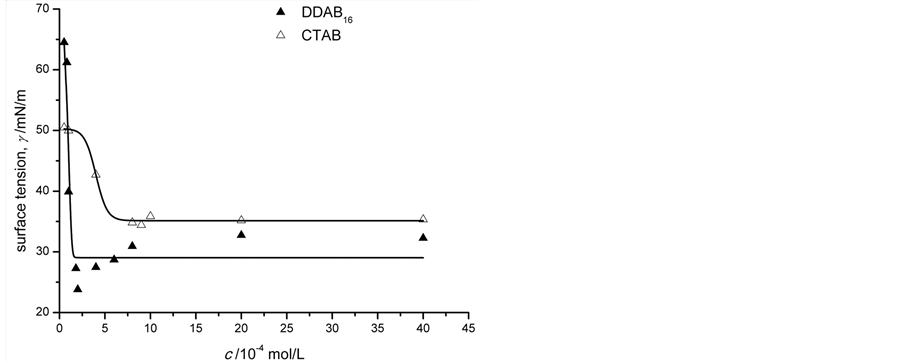
Figure 1. Effect of surfactant concentration on the surface tension in aqueous solution
图1. 表面活性剂浓度对表面张力的影响
带负电荷的亲水表面层和Tween-80中氧原子带有的电子对,都对带负电荷的钌催化剂活性物种具有排斥作用,使胶束亲水表面层中钌催化剂活性物种浓度明显降低,不利于底物与催化剂间的配位,因而降低了反应速率。
3.2. 双长链表面活性剂泡囊体系中催化剂的循环使用
表2为在水–有机两相体系中,双长链表面活性剂存在下进行苯乙酮加氢反应时,催化剂的循环反应
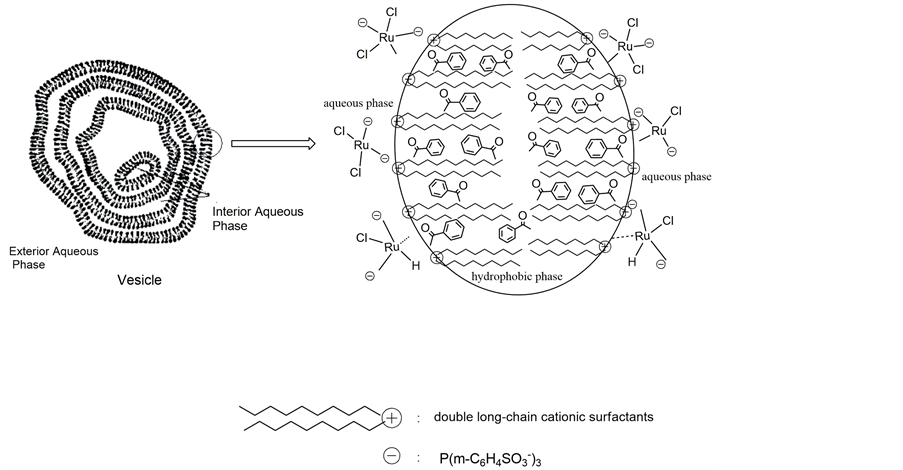
Figure 2. Sketch of vesicle formed by double long-chain cationic surfactants
图2. 双长链表面活性剂形成的泡囊结构胶束示意图
Table 2. Recycling and reuse of catalyst in vesicle system
表2. 催化剂的循环使用
aThe reaction conditions are the same as that listed in Table 1. b0.5 mL 2-propanol was added in every recycle time. The other reaction conditions are the same as that listed in Table 1. c0.0025 mmol TPPTS and 0.5 mL 2-propanolwere added in every recycle time. The other reaction conditions are the same as that listed in Table 1. d0.0025 mmol TPPTS and 0.5 mL 2-propanol were added in the fifth recycle time. The other reaction conditions are the same as that listed in Table 1. e0.0025 mmol TPPTS were added in the fifth recycletime. The other reaction conditions are the same as that listed in Table 1.
实验结果。在第一组循环实验中(Entry1),每次反应完成后让溶液静置分层,分离出有机层后,立即将反应物加入催化剂水溶液中继续进行第二次反应,循环四次苯乙酮的转化率从82.3%降低至41.0%。引起这一结果的原因可能有两个:1) 氢转移反应中提供氢源的异丙醇会随着分出有机层时被带走,使催化剂水溶液中氢转移试剂浓度越来越低;2) 进行分离时接触到空气会使少量膦配体TPPTS被氧化。因而,尝试了在第五次循环时添加了少量异丙醇和TPPTS,苯乙酮转化率立即提高到75.1%。
为了确认上面分析的两个因素对反应转化率的影响,进行了第二组循环实验(Entry2)。在每次反应结束分离出有机层后,加入底物的同时补加少量异丙醇,当反应循环至第四次的时候,转化率降低到72.4%,这明显地比第一组循环实验的转化率降低小很多。此时,如果在下一次循环前,补加适量的TPPTS,转化率恢复到接近第二次循环的水平。这个结果说明,在以异丙醇为氢源的水–有机两相体系中,每次分离有机相带走异丙醇对循环反应转化率的影响是主要的,同时,多次循环分离操作与空气的接触,使得膦配体少量氧化也会影响氢转移反应的转化率。因此,在催化剂的循环实验中,每次循环实验前都要补加适量的TPPTS和异丙醇,此时催化剂循环使用10次的情况下,转化率仍保持在比较高的水平(entry 3)。
4. 结论
研究结果表明,在水–有机两相体系中苯乙酮催化转移氢化反应,双长链阳离子表面活性剂的加入可以有效地提高反应速度。这是由于双长链表面活性剂在水相中形成的泡囊结构造成的反应微环境有利于苯乙酮与钌催化剂活性物种的配位反应;同时,在水–有机两相体系中,水溶性催化剂与产物容易分离,有利于催化剂的再循环使用。
致谢
感谢国家自然科学基金项目(No.21371079)、嘉兴学院大学生研究训练(SRT)计划项目(批准号:851714062)对本工作的支持。
文章引用
龙嘉豪,方 权,刘益美,张雨婷,马红霞, (2015) 水相体系中苯乙酮催化氢转移制备苯乙醇
Catalyzed Transfer Hydrogenation of Acetophenone to Phenethyl Alcohol in Water. 合成化学研究,01,1-6. doi: 10.12677/SSC.2015.31001
参考文献 (References)
- 1. Mizushima, E., Ohi, H., Yamaguchi, M., et al. (1999) Asymmetric transfer hydrogenation of aryl-alkyl ketones catalyzed by Ruthenium(II) complexes having chiral pyridylmethylamine and phosphine ligands. Journal of Molecular Catalysis A: Chemical, 149, 43-49.
- 2. Gao, J.X., Yi, X.D., Xu, P.P., et al. (1999) New chiral cationic rhodium-aminophosphine complexes for asymmetric transfer hydrogenation of aromatic ketones. The Journal of Organic Chemistry, 592, 290-295.
- 3. Hashiguchi, S., Fujiii, A., Takehara, J., et al. (1995) Asymmetric transfer hydrogenation of aromatic ketones catalyzed by Chiral Ruthenium(II) Complexes. Journal of the American Chemical Society, 117, 7562-7563.
- 4. Noyori, R. and Hashiguchi, S. (1997) Asymmetric transfer hydrogenation catalyzed by chiral Ruthenium complexes. Accounts of Chemical Research, 30, 97-102.
- 5. Wu, X., Li, X., Hems, W., et al. (2004) Ac-celerated asymmetric transfer hydrogenation of aromatic ketones in water. Organic & Biomolecular Chemistry, 2, 1818-1821.
- 6. Wu, X., Wang, C. and Xiao, J. (2010) Asymmetric transfer hydrogenation in water with platinum group metal catalysts. Platinum Metals Review, 54, 3-19.
- 7. Sun, Y., Liu, G., Gu, H., et al. (2011) Magnetically re-coverable SiO2-coated Fe3O4 nanoparticles: A new platform for asymmetric transfer hydrogenation of aromatic ketones in aqueous medium. Chemical Communications, 47, 2583- 2585.
- 8. Huo, H., Zhou, Z., Zhang, A., et al. (2012) Ru-thenium(II)-catalyzed transfer hydrogenation of aldehydes with new water-soluble monotosylated ethylenediamines as ligands. Research on Chemical Intermediates, 38, 261-268.
- 9. Chaudhari, R.V., Bhanage, B.M., Deshpande, R.M., et al. (1995) Enhancement of interfacial catalysis in a biphasic system using catalyst-binding ligands. Nature, 373, 501-503.
- 10. Cornils, B. and Herrmann, W.A. (2006) Aqueous-phase organometallic catalysis. Wiley-VCH, Wein-heim.
- 11. Dwars, T., Paetzold, E. and Oehme, G. (2005) Reactions in micellar systems. Angewandte Chemie Interna-tional Edition, 44, 7174-7199.
- 12. Li, J., Zhang, Y.M., Han, D.F., Jia, G.Q., Gao, J.B., Zhong, L., et al. (2008) Transfer hydrogenation of aldehydes on amphiphilic catalyst assembled at the interface of emulsion droplets. Green Chemistry, 10, 608-611.
- 13. Schwarze, M., Milano-Brusco, J.S., Strempel, V., Hamerla, T., Wille, S., Fischer, C., et al. (2011) Rhodium catalyzed hydrogenation reactions in aqueous micellar systems as green solvents. RSC Advances, 1, 474-483.
- 14. Kunishima, M., Kikuchi, K., Kawai, Y. and Hioki, K. (2012) Substrate-Selective dehydrocondensation at the interface of micelles and emulsions of common surfactants. Angewandte Chemie International Edition, 51, 2080-2083.
- 15. Li, J.H., Li, X.F., Ma, Y.P., Wu, J.S., Wang, F., Xiang, J., et al. (2013) Surfactant-Accelerated asymmetric transfer hydrogenation with recyclable water-soluble catalyst in aqueous media. RSC Advances, 3, 1825-1834.
- 16. Stephenson, T.A. and Wilkinson, G.J. (1966) New complexes of Ruthenium (II) and (III) with tri-phenylphorostannate, pyridine and other ligands. Journal of Inorganic and Nuclear Chemistry, 28, 945-956.
- 17. Fendler, J.H. (1980) Microemulsions, micelles, and vesicles as media for membrane mimetic photo-chemistry. The Journal of Physical Chemistry, 84, 1485-1491.
- 18. Goedheijt, M.S., Hanson, B.E., Reek, J.N.H., Ka-mer, P.C.J. and van Leeuwen, P.W.N.M. (2000) Accelerated biphasic hydroformylation by vesicle formation of am-phiphilic diphosphines. Journal of the American Chemical Society, 122, 1650-1657.