Pharmacy Information
Vol.
12
No.
02
(
2023
), Article ID:
63704
,
19
pages
10.12677/PI.2023.122018
ATR小分子抑制剂的研究进展
程浩东,段云鑫,施志浩*
中国药科大学理学院,江苏 南京
收稿日期:2023年2月22日;录用日期:2023年3月23日;发布日期:2023年3月31日

摘要
共济失调毛细血管扩张和Rad3相关激酶(ATR)是DNA损伤反应(DDR)的重要调节因子,尤其是对复制压力(RS)的反应。由于DNA损伤和RS是基因组不稳定性的主要来源,选择性抑制ATR是癌症治疗的一种有前途的新方法。本文综述了ATR的结构功能以及ATR小分子抑制剂的研究进展,以求为此领域的进一步研究工作提供理论帮助和指导。
关键词
DNA损伤反应,ATR激酶,抑制剂,抗肿瘤

Advances in the Study of Small Molecule Inhibitors of ATR
Haodong Cheng, Yunxin Duan, Zhihao Shi*
School of Science, China Pharmaceutical University, Nanjing Jiangsu
Received: Feb. 22nd, 2023; accepted: Mar. 23rd, 2023; published: Mar. 31st, 2023

ABSTRACT
Ataxia-telangiectasia and Rad3-related kinase (ATR) are important regulators of the DNA damage response (DDR), particularly in response to replication stress (RS). As DDR and RS are major sources of genomic instability, selective inhibition of ATR is a promising new approach for cancer therapy. This article reviews the structural function of ATR and the progress of research on small molecule inhibitors of ATR in order to provide theoretical assistance and guidance for further research in this field.
Keywords:DNA Damage Response, ATR Kinase, Inhibitor, Antitumour

Copyright © 2023 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/


1. 概述
基因组的完整性对于生物体的生存至关重要,但DNA不断受到各种外部和内部影响,导致DNA丢失,如碱基错配、DNA单/双链断裂损伤(single/double-strand break damage, S/DSB)、链间或链内交联等 [1] [2] [3] 。同时,细胞对不同的损伤有一系列的修复机制。DNA损伤修复是一个严格控制的复杂信号网络,也被称为DNA损伤反应(DNA damage response, DDR)。DDR通路是一种参与细胞DNA损伤检测、细胞周期适应和损伤修复过程的复杂网络。当DDR通路存在缺陷或失调时可以促进肿瘤的发生,同时可能增加癌细胞对替代修复途径的依赖。这些网络在正常细胞和癌细胞中都发挥着关键作用,以维持细胞活力和基因组稳定性 [4] 。DDR通路不断监测DNA的完整性,在出现任何类型的DNA损伤时,激活短暂的细胞周期停滞来修复DNA,以确保维持基因组的稳定性和细胞的生存能力 [5] 。因此,DDR对维持基因组的完整性及生物体的生存都至关重要。
磷脂酰肌醇3-激酶样激酶(phosphatidylinositol 3-kinase like kinase, PIKK)属于丝氨酸/苏氨酸激酶家族,该家族参与一系列不同的细胞功能,包括减数分裂、DNA损伤感应和修复、细胞存活、增殖和细胞生长等。其中,共济失调毛细血管扩张和Rad3相关激酶(ataxia-telangiectasia and Rad3-related kinase, ATR)属于PIKK蛋白家族的一种,是DDR通路中的关键调控因子 [6] [7] 。该家族的其他成员还包括共济失调毛细血管扩张突变(ataxia telangiectasia mutated, ATM),DNA依赖性蛋白激酶(DNA-dependent protein kinase, DNA-PK),雷帕霉素的哺乳动物靶点(mammalian target of rapamycin, mTOR)等。ATR通过激活DNA损伤修复的必要信号通路,特别是在复制压力(replication stress, RS)反应中,在稳定复制叉、调节细胞周期进程和激活DNA损伤修复中均发挥着关键作用 [8] [9] [10] 。
2. ATR结构与功能
ATR是一种301.66 kDa的蛋白质,于1996年作为酵母中有丝分裂进入检查点蛋白1 (mitosis entry checkpoint protein 1, Mec1)的哺乳动物直系同源物中首次被发现 [11] 。ATR的C端激酶结构域的结构上与PIKK激酶家族其他成员表现出相当大的相似性,可磷酸化下游蛋白。N端包含ATR相互作用蛋白(ATR-interacting protein, ATRIP)的结合位点,ATRIP负责调节ATR在复制压力和DNA损伤位点的定位。在单链DNA (single-stranded DNA, ssDNA)的位点上,复制蛋白A (replication protein A, RPA)包被的ssDNA片段激活ATR,RPA与ssDNA结合并覆盖,以保护其免于进一步降解。然后,通过RPA和ATRIP之间的直接相互作用,使ATR得以定位,其对ATR信号传递至关重要。
当ATR检测到持久性单链DNA (single-stranded DNA, ssDNA)的存在时,它会被激活,然后触发DNA修复过程。首先,包被在ssDNA上的复制蛋白A (replication protein A, RPA)与ATRIP结合,后者会将ATR募集到靠近三聚体Rad9-Rad1-Hus1复合物(9-1-1)的位点。随后ATRIP-ATR复合物会磷酸化9-1-1。磷酸化的9-1-1与拓扑异构酶结合蛋白1 (topoisomerase-binding protein 1, TOBP1)结合以激活ATR信号级联。抑制ATR会干扰下游信号转导检查点激酶1 (checkpoint kinase 1, CHK1),从而导致细胞凋亡(图1)。此外,由于染色质过早凝结和有丝分裂的进入,ATR还可以通过磷酸化p53肿瘤抑制因子,导致p53的凋亡 [12] 。
与家族其他成员功能不同,ATR主要负责ssDNA损伤响应,更具体地说是复制压力(replication stress, RS)。DNA复制压力是癌症产生的一个标志,ATR通过稳定复制叉、调节细胞周期进程和激活DNA损伤修复,在DNA复制应激反应中起着关键作用。大量研究表明,当ATM功能丧失(如通过Ras激活或MYC扩增诱导癌基因)或RS导致的DDR缺陷(如ATM功能丧失)时,癌细胞通常更依赖ATR通路来生存 [13] 。这提供了利用ATR抑制剂杀死癌细胞并且保留正常非癌细胞的可能性。选择性ATR抑制可能增强对某些DNA损伤诱导治疗的敏感性,以及对某些DNA损伤修复的损害治疗,为ATR抑制剂在抗癌治疗中提供了强大的潜力。这些研究结果进一步推动了开发ATR抑制剂的研究。目前,还缺乏有效的ATR抑制剂。因此,需要新的化学实体,可以选择性地抑制ATR,用于临床使用或进一步推动ATR抑制剂的研究。

Figure 1. ATR’s roles in DDR pathway [14]
图1. ATR在DDR通路中的作用 [14]
3. ATR小分子抑制剂的研究进展
近十年,ATR靶标得到了众多医药企业和研究机构的青睐,默克、阿斯利康、拜耳、修复治疗、恒瑞医药、英派药业和德琪医药等制药公司通过自主开发、产品收购等方式推出了一系列ATR小分子抑制剂,目前有10多种小分子抑制处于不同临床研究阶段,具有代表性的ATR抑制剂如下表所示(见表1)。
目前,尚无ATR抑制剂被批准上市,已报道的部分ATR抑制剂按结构可大致分为:吡嗪类、嘧啶类、吡啶类、稠环嘧啶类、稠环吡啶类和其他类等。本文按照大致的结构类型,介绍近十年有代表性的抑制剂的设计、结构改造策略和抗肿瘤活性研究,为研究人员设计新型ATR抑制剂提供改造思路。
1. 吡嗪类
沃泰克斯(Vertex)制药公司于2011年报道了首个高活性ATR抑制剂VE-821 (见图2),其对ATR的激酶抑制IC50值为26 nM,但细胞活性和理化性质仍需改善。因考虑到分子结构中的苯基酰胺部分可能

Table 1. Part of ATR inhibitors and related clinical information
表1. 部分 ATR抑制剂及相关临床信息
*AST:晚期实体瘤;HNSCC:头颈鳞癌;OC:卵巢癌;mCRPC:转移性去势抵抗性前列腺癌;UC尿路上皮癌;SCLC:小细胞肺癌;NSCLC:非小细胞肺癌;CLL:慢性淋巴细胞白血病;NHL:非霍奇金淋巴瘤;MDS:骨髓增生异常综合征;CMML:慢性粒单核细胞白血病;BDC:胆管癌;PC:胰腺癌;TNBC:三阴性乳腺癌;MC:转移癌;PPC:原发性腹膜癌;FTC:输卵管癌。
产生毒性代谢产物,研究人员首先对酰胺连接子进行了优化,采用了多种五元杂环作为连接子,以异噁唑为连接子的化合物2的活性相较于化合物1有了很大的提升,且避免了毒性代谢产物的产生。对占据后口袋的苯环继续进行结构优化得到化合物3 (见图2),其对ATR激酶抑制Ki为0.17 nM。对同家族的ATM和DNA-PK的Ki值分别为44和大于4000 nM,同时也表现出很强的细胞活性和良好的代谢性质和安全性。分析其与ATR蛋白结构的结合模式,M6620分子呈现V型构象与蛋白结合,分子结构中的氨基吡嗪片段与铰链区Val2380和Glu2378形成双齿氢键,异丙砜基苯基中氧原子同Gly2385产生氢键相互作用。此外,苄胺片段中的NH同Asp2494和Glu2479残基各形成一处氢键,该相互作用的存在可能是M6620较化合物2活性提升的原因之一 [15] 。M6620是首个进入临床研究的ATR小分子抑制剂,目前处于临床I/II期研究阶段。
2. 嘧啶类
阿斯利康(AstraZeneca)制药公司从化合物库中筛选得到嘧啶类先导化合物4 (见图3),其对ATR的激酶抑制IC50值为30 nM,在HT29人结肠癌细胞中,对ATR下游Chk1蛋白Ser-345的磷酸化抑制IC50值为1.1 μM。研究人员对化合物4与PI3Kγ的结合模式进行了分析,分子结构中吗啉的氧原子同铰链区Val2380形成关键氢键,吲哚片段占据激酶后口袋位置,乙砜基则伸向溶剂域。对化合物4进行初步优化得到了化合物5 (见图3),激酶活性和细胞活性都得到了显著提升(ATR IC50 = 5 nM,ATR cell IC50 = 50 nM)。后续研
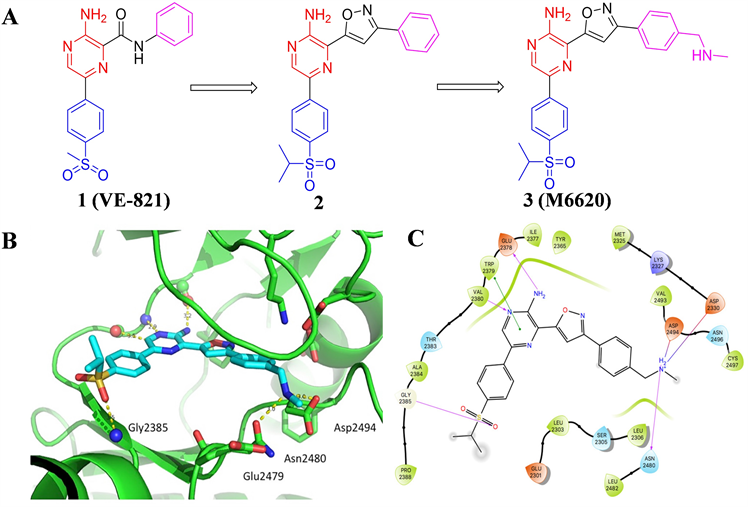
Figure 2. (A) The chemical structures of compound 1, 2 and 3 (The red part of structure represents nucleus of molecule. The purple part represents fragment occupies back pocket. The purple part represents fragment orient to solvent region, the same below). (B) M6620 docked into the active site of ATR. (PDB: 1E7V). (C) Ligand interaction diagram showing nearby residues involving M6620 as docked into ATR. (Aromatic stacking (green lines), key hydrogen bonds (purple lines)) [15]
图2. (A) 化合物1、2和3的化学结构(结构中红色部分表示分子的母核,紫色部分表示占据后口袋的结构片段,蓝色部分表示朝向溶剂域的结构片段,下同);(B) M6620与ATR活性位点的结合模式(PDB:1E7V);(C) M6620与附近残基产生的相互作用图(绿线表示芳香堆积作用,紫线表示关键氢键作用) [15]
究中,在分子的吗啉基团中引入R构型甲基对化合物的活性及选择性提升有利,因此后续开发的很多ATR小分子药物中基本都采用了(R)-3-甲基吗啉这一结构片段。在小鼠LoVo结肠癌异种移植瘤模型中,50 mg/kg每天给药一次能够实现明显的肿瘤抑制,TGI为77%。AZ20虽表现出不错的体内外活性,但仍存在一些成药性方面的问题,例如水溶性差,能够对CYP3A4酶产生时间依赖性抑制等。针对这些问题,研究人员对AZ20进行进一步的结构优化得到化合物6 (见图3),其中将砜基替换为亚砜亚胺基团,显著改善了化合物的水溶性,将吲哚片段替换为氮杂吲哚片段,降低了化合物对CYP3A4酶产生的时间依赖性抑制。分析AZD6738的结合模式,(R)-3-甲基吗啉上的氧原子和氮杂吲哚基团的NH分别于激酶铰链区和后口袋氨基酸残基产生关键的氢键相互作用,亚砜亚胺朝向溶剂域。AZD6738单药对一系列对携带DNA损伤修复缺陷的肿瘤具有良好的体内外活性,并且能够与多种作用机制不同的抗肿瘤药物组成联合疗法在多种类型的肿瘤具有治疗潜力 [16] 。目前阿斯利康针对AZD6738已经开展了多项单药或联合用药的临床研究,目前处于临床I/II期。
2019年至2021年间,美国德州大学系统、北京泰德制药和石家庄智康弘仁公司先后公开了数篇专利,相关化合物结构均是在AZD6738的基础上,对分子中的亚砜亚胺基团和氮杂吲哚基团进行结构改造的mee-too类药物专利 [17] [18] 。其中德州大学系统两篇专利中的代表性化合物分别采用二甲基亚磺酰亚胺和环丙磺酰基哌啶基结构替换AZD6738结构中的环丙基亚砜亚胺片段,得到代表性化合物7和8 (见

Figure 3. (A) The chemical structures of compound 4-6. (B) Molecular docking model of compound 4 with PI3Kγ. (PDB: 3DBS). (C) Molecular docking model of compound 6 with ATR. (PDB: 5UK8)
图3. (A) 化合物4-6的化学结构;(B) 化合物4与PI3Kγ的结合模式(PDB:3DBS) [7] ;(C) 化合物6与ATR的结合模式(PDB: 5UK8) [16]
图4)。其中化合物7对ATR的激酶抑制IC50值为1 nM,对Chk1的磷酸化抑制IC50为46 nM。在小鼠LoVo结肠癌异种移植瘤模型,100 mg/kg 剂量给药即可实现96%肿瘤抑制率,并且给药期间,小鼠体重没有出现明显下降,体现了良好的安全性。化合物8同样表现出良好的激酶和细胞抑制活性(ATR IC50 = 5 nM, ATR cell IC50 = 21 nM),但这两类小分子的后续进展没有进一步的报道。北京泰德制药公开的专利中,代表化合物9 (见图4)采用N-甲基-1H-苯并[d]咪唑-2-胺结构替换AZD6738分子结构中的氮杂吲哚片段,并在嘧啶环的4位引入4-(5-甲基-3-(三氟甲基)-1H-吡唑-1-基)哌啶片段,其对ATR的IC50为3 nM,对卵巢癌TOV21G和白血病MV4-11细胞的抗增殖IC50分别为0.12和0.08 μM。化合物9在人和小鼠的肝微粒体中表现出较长的半衰期,分别为203和110分钟。化合物9在小鼠体内也表现优良的药代动力学性质,口服T1/2为3.1小时,Cmax为467 ng/mL,AUC为5452 h*ng/mL,但未见体内药效学数据报道。
化合物10 (见图4)公开于石家庄智康弘仁公司申请的专利。其分子结构中1,4-二甲基-1H-吡唑片段占据亲水区,并采用6-羟甲基吲哚片段占据后口袋。化合物10的激酶和细胞抗增殖活性良好(ATR IC50 = 29 nM,LoVo IC50 = 0.51 μM),小鼠药代动力学性质优异(口服T1/2 = 2.02 h,Cmax = 6500 nM,AUC = 5681 nM*h,F = 129.0 %),在小鼠结直肠癌LoVo CDX体内药效研究中,给药剂量为25mg/kg,每天给药两次,肿瘤生长抑制率达到90.7 %,表现出优异的体内疗效 [19] 。
3. 吡啶类
德州大学系统、德琪医药和深圳瓴方生物报道了三篇吡啶类化合物作为ATR抑制剂的专利 [20] [21] [22] 。分析专利包含的化合物和活性数据,推测AZD6738的嘧啶母核不一定是分子维持ATR活性所必需的结构,可以使用吡啶环替代。
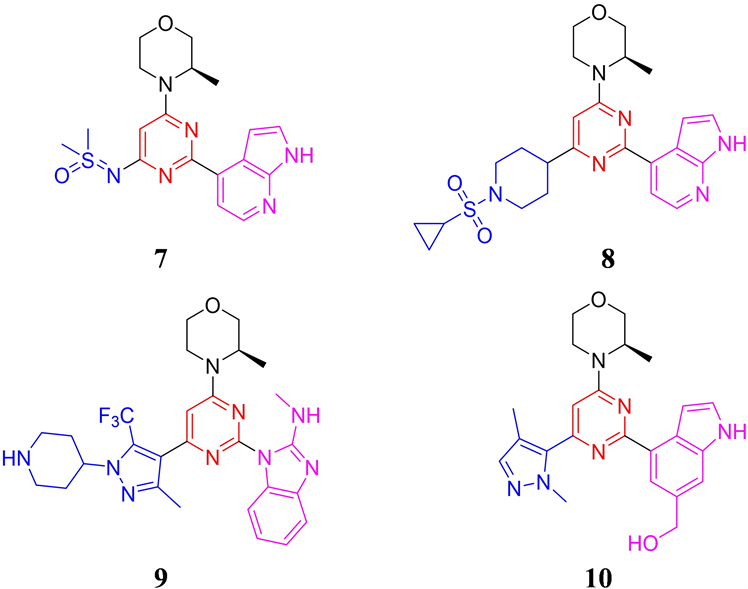
Figure 4. The chemical structures of compound 7-10
图4. 化合物7-10的化学结构
例如美国德州大学公开专利中的代表化合物11 (见图5)可强烈抑制ATR激酶活性,IC50值为2 nM,并能强效抑制胞内下游Chk1的磷酸化水平,IC50值为44 nM。德琪医药的专利中的化合物12 (见图5),除了采用吡啶核心外,还将伸向后口袋的氮杂吲哚结构替换为氨基吡唑片段,其依然保持了良好的ATR激酶抑制和细胞活性(ATR IC50= 14.4 nM, LoVo IC50= 334 nM),深圳翎方生物公开的专利中,代表化合物13则在采用吡啶为母核的同时,将伸向溶剂域的片段替换为1-甲基-1H-1,2,3-三氮唑片段。化合物13对ATR激酶和LoVo结肠癌细胞均表现出良好抑制,IC50值分别为33和103 nM,且PK性质良好(口服T1/2 = 1.4 h,Cmax = 9565 nM,AUC = 22992 nM*h,F = 91.6 %)。在人胃癌细胞SNU-601异种移植瘤模型中研究化合物13的抗肿瘤体内药效,给药剂量为15 mg/kg,每周给药3天后停药4天,抑瘤率达到104.8%,且实验期间给药组小鼠体重无明显下降,动物状态无异常。表明化合物13能够抑制人胃癌SNU-601异种移植瘤的生长,且小鼠相对耐受。
4. 稠环嘧啶类
四川大学杨胜勇课题组以从化合物库中筛选得到的以喹唑啉为母核的先导化合物14为基础,分别对分子结构中的苯基、(R)-3-甲基吗啉和吲哚基团进行了深入的构效关系研究,得到最优化合物15 (SKLB-197) (见图6) [23] 。SKLB-197对ATR的激酶抑制IC50为13 nM。在对402种激酶的筛选中,

Figure 5. The chemical structures of compound 11-13
图5. 化合物11-13的化学结构
SKLB-197对ATR表现出高度选择性,仅对B-Raf激酶产生微弱抑制活性,IC50值为1.774 μM。分子模拟结果显示,SKLB-197分子中的(R)-3-甲基吗啉结构中氧原子与铰链区Val851残基产生关键氢键,伸向后口袋的吲哚基团与Leu807、Ile932和Phe934产生疏水相互作用,吲哚结构中的NH与疏水区的Asp810残基产生氢键相互作用。甲基吡唑结构的N原子与Gln859残基形成氢键,甲基则伸向由Trp850和Met922残基构成的小疏水空腔,这种相互作用限制了SKLB-197的分子构象,是导致化合物对ATR表现出高选择性的潜在原因。蛋白免疫印迹结果显示,SKLB-197能够剂量依赖性地抑制ATR及其下游Chk1蛋白的磷酸化水平。细胞抗增殖测试结果表明,SKLB-197对于包括LoVo、Granta-519和OVCAR-3等在内多种携带ATM缺陷的细胞株表现强烈的抑制活性。对ATM表达正常的细胞株则不产生明显抑制。SKLB-197联合ATM抑制剂AZD1390给药,可显著抑制ATM表达正常的HT-29细胞株的增殖,并通过诱导胞内DNA损伤水平增加促使细胞凋亡。SKLB-197在大鼠体内具有优越的药代动力学性质,进而进行了体内抗肿瘤活性研究。在LoVo结肠癌小鼠异种移植瘤模型中,给药剂量100 mg/kg,每天给药两次,给药30天,肿瘤生长抑制率达到72.8%,且给药期间小鼠体重没有明显变化,表明SKLB-197良好的体内药效和安全性。
2022年5月,四川大学的Li等发现了一类以6,7-二氢-5H-吡咯并[3,4-d]嘧啶为母核的ATR小分子抑制剂,其中代表性化合物20 (见图7)对ATR表现出纳摩尔级别的抑制活性,IC50值为7 nM [24] 。测试化合物20对多种携带DNA损伤修复缺陷细胞株的抗增殖活性,结果显示,A2780卵巢癌细胞对化合物16 (见图7)最为敏感,增殖抑制IC50值为0.67 μM。此外,研究人员通过克隆形成和划痕实验,证明化合物16能够抑制A2780细胞克隆形成和迁移能力。蛋白免疫印迹实验结果显示,在A2780细胞中,化合物16能够剂量依赖性地抑制ATR及其下游Chk1蛋白的磷酸化,诱导胞内DNA产生损伤,上调胞内γH2AX水平。此外,化合物16也在402种激酶的筛选中表现优良的选择性,仅对mTOR和mTOR/FKBP12产生中等程度抑制,IC50值分别为0.136和0.125 μM。
恒瑞医药公开了一类以咪唑并[1,5-a]嘧啶为母核的小分子抑制剂,其中代表化合物17 (见图7)对ATR激酶表现显著和抑制活性和选择性(ATR IC50 = 8 nM, ATM IC50 > 10000 nM, DNA-PK IC50 = 663 nM, PI3K IC50 = 5131 nM),对LoVo细胞具有优越的抗增殖活性(LoVo IC50 = 47 nM),同时具有良好的药代动力学性质 [25] 。目前该类化合物的后续进展还有待公开。
此外,恒瑞公司还先后公开了6,7-二氢-5H-吡咯并[2,3-d]嘧啶和5,7-二氢噻吩[3,4-d]嘧啶-6,6-二氧化物两类母核化合物的专利,代表化合物18和19 (见图7)对ATR虽表现出良好的抑制活性和选择性,但细胞活性一般,对LoVo细胞的抗增殖活性仅为数百纳摩尔 [26] [27] 。
上海迪诺医药公开了一类以7,8-二氢-6H-吡喃并[3,2-d]嘧啶为母核的小分子抑制剂,代表化合物20经酶学测试,对ATR的IC50值为1.75 nM。细胞活性结果表明,化合物20能够强烈抑制LoVo细胞增殖,IC50值为15.26 nM。同时,研究人员也对其进行了pChk1细胞水平检测,IC50值11.1 nM。该专利还包含了数个活性突出的类似化合物结构,具有一定的临床潜力 [28] 。
默克公司公开了一类嘧啶并噁嗪类衍生物作为ATR抑制剂的专利,研究人员巧妙地将嘧啶母核同(R)-3-甲基吗啉结构进行环合,得到了一类以化合物21 (见图7)为代表结构新颖的三环类化合物,但专利并未揭示化合物具体的活性数据。
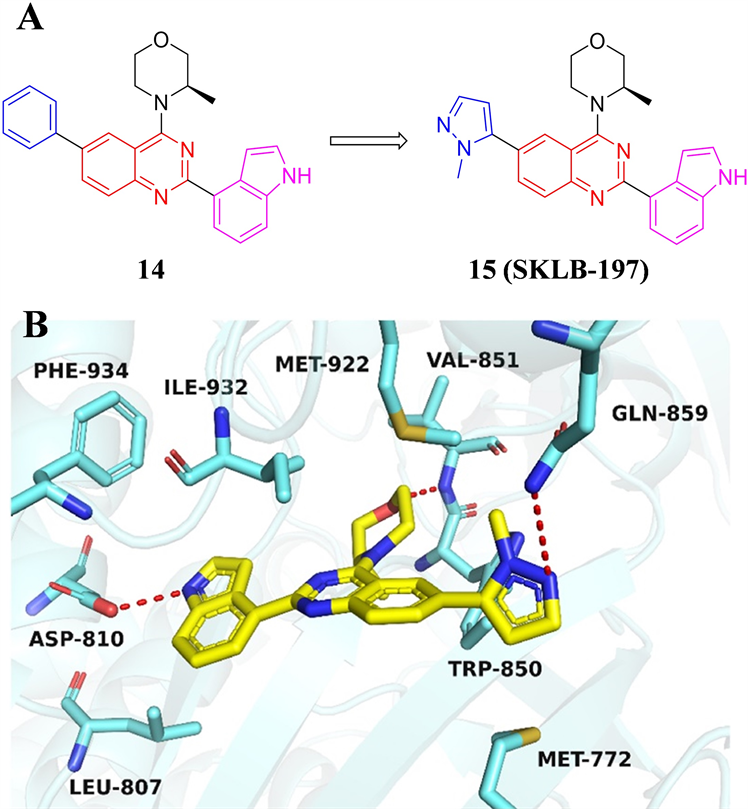
Figure 6. (A) The chemical structures of compound 14 and 15. (B) Molecular docking model of compound 15 with ATR (PDB: 5UK8)
图6. (A) 化合物14和15的化学结构;(B) 化合物15与ATR的结合模式(PDB: 5UK8)

Figure 7. The chemical structures of compound 16-21
图7. 化合物16-21的化学结构
5. 稠环吡啶类
拜耳公司通过高通量筛选技术得到了喹啉类苗头化合物22 (见图8),考虑到分子结构中的富电子吡咯环会带来DMPK不佳的问题,并兼顾化学合成的简便性,对分子结构进行初步优化,得到了一类以1,7-萘啶结构的新型ATR小分子抑制剂化合物23 (BAY-937),其对ATR激酶的抑制IC50值为59 nM,但对mTOR的选择性不高,且水溶性和PK性质一般。考虑到脱靶毒性和成药性问题,研究人员对化合物23进行进一步优化,将吗啉替换为(R)-3-甲基吗啉结构,改善了化合物的活性及选择性,同时将对甲砜基苯基替换为甲基吡唑基团,进一步优化化合物的结构得到了化合物24 (BAY 1895344)。BAY 1895344对于ATR激酶及多种细胞株均表现强烈的抑制活性,对mTOR激酶也实现了超过60倍的选择性,良好的活性和成药性使得该化合物在多种类型的移植瘤模型中展现出优异的体内药效和安全性 [29] [30] 。目前该化合物正处于对晚期实体瘤和淋巴瘤患者治疗的临床I期研究中 [31] 。对化合物24的结合模式进行分析,其分子结构中(R)-3-甲基吗啉上的O原子和吡唑结构的NH分别于激酶铰链区的Val851和后口袋Asp810产生两处关键的氢键相互作用,甲基吡唑基团朝向溶剂域。我们对化合物24和AZD6738进行分子叠合,发现二者表现相似的结合模式,将AZD6738的嘧啶母核的2、3位进行6元环的环合,并将氮杂吲哚结构进行开环,经过结构优化即得到BAY 1895344。
BAY 1895344的体内外活性突出,构效关系显示,BAY 1895344结构中的(R)-3-甲基吗啉和占据后口袋的吡唑结构对保持分子活性至关重要,因此1,7-萘啶母核以及朝向溶剂域的甲基吡唑结构成为了研究人员结构优化的重点。科伦药业的公开了一类吡啶并[3,4-b]吡嗪-2(1H)-酮为骨架的ATR抑制剂,代表化合物25 (见图9)对ATR激酶和肺癌NCI-H23细胞具有中等程度的抑制活性,IC50值分别为0.147和0.020 μM [32] 。默克公司则采用了五元并六元环作为母核,代表化合物26表现纳摩尔级别的激酶抑制活性 [33] ,而以1H-吡唑并[4,3-b]吡啶的核心的化合物在后续恒瑞医药和贝达药业公开的ATR抑制剂专利中也有涉及 [34] [35] 。德琪医药在2022年公开的专利中,采用了异噻唑并[4,5-b]吡啶母核,代表化合物27对ATR的激酶抑制IC50小于100 nM,对LoVo细胞的增殖抑制IC50小于1 nM [36] 。英派药业和北京泰德则分别报道了以吡唑并[1,5-a]嘧啶和咪唑并[1,5-b]哒嗪为母核的ATR抑制剂结构,代表化合物28和29均具有良好的体外活性 [37] [38] 。修复治疗公司报道了一类吡唑并[5,4-b]吡啶类ATR小分子抑制剂,代表化合物30 (RP-3500)其ATR的激酶 IC50为 0.9 nM,且具有高度的选择性,对多种携带DDR缺陷的肿瘤细胞株具有优异的抗增殖活性,单药和联合用药均能在多种移植瘤模型中展现良好的抑瘤效果 [39] 。RP-3500目前已进入临床研究,正在评估其在携带DNA损伤修复缺陷的晚期实体瘤中的安全性和有效性。上海迪诺则另辟蹊径,在RP-3500的结构的基础上通过合环策略得到了一类以1,2,3,6-四氢吡唑并[3,4-b]吡咯并[2,3-d]吡啶作为全新母核的ATR小分子抑制剂,代表化合物31 (见图9)的安全性好、体外活性突出(ATR IC50= 0.69 nM,LoVo IC50= 37.34 nM),同时在OCI-LY19人B细胞淋巴瘤小鼠皮下移植瘤模型中,给药剂量为10 mg/kg,每天给药一次,给药14天时,抑瘤率达到95.9%,优于临床在研化合物AZD6738 (给药剂量20 mg/kg,TGI为72.1%),该化合物有一定的临床潜力 [40] 。南京明德新药报道的专利显示,其分子结构依然采用了1,7-萘啶母核,但在5位引入了F原子,因对分子改动较小,代表化合物32基本保持了BAY1895344体外活性,但F原子的引入优化了PK性质,这使得化合物33在LoVo细胞异种移植瘤模型中,能以更低的口服剂量(化合物33:40 mg/kg vs BAY 1895344: 50mg/kg)实现优良的肿瘤生长抑制效果 [41] 。

Figure 8. (A) The chemical structures of compound 22-24. (B) Molecular docking model of compound 24 with ATR (PDB: 5UK8). (C) Superimposition of 24 with AZD6738 in ATR
图8. (A) 化合物22-24的化学结构;(B) 化合物24与ATR的结合模式(PDB: 5UK8);(C) 化合物24与AZD6738的叠合
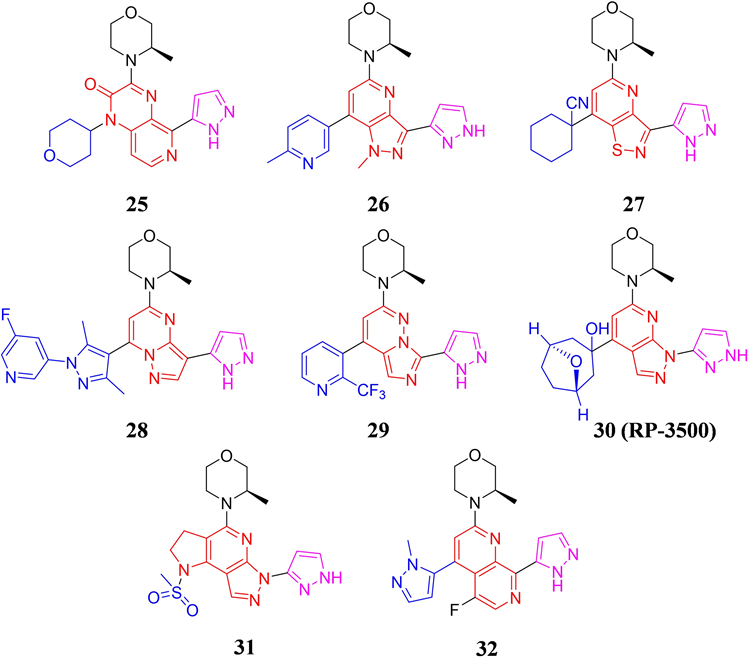
Figure 9. The chemical structures of compound 25-32
图9. 化合物25-32的化学结构
6. 稠环吡唑类
默克公司先后从Vertex制药公司收购了ATR小分子抑制剂M4344 (化合物33)和M1774 (化合物34) (见图10),两个小分子具有类似的化学结构,核心为6-氟吡唑并[1,5-a]嘧啶-2-胺,其中,M4344对ATR的Ki值小于0.15 nM,可高效抑制ATR驱动的Chk1的磷酸化,IC50值为8 nM。M4344能够与临床上广泛使用的化疗药物,如依托泊苷、吉西他滨、顺铂等联用,通过协同作用增强药物的治疗效果 [42] 。M1774也是一种高效、选择性的口服ATR抑制剂,在多种人源和对ATR抑制剂敏感的AML异种移植瘤模型中均能展现优越的抗肿瘤活性 [43] 。Vertex公司在2014-2015年间公开了两篇ATR抑制剂专利,均是在6-氟吡唑并[1,5-a]嘧啶-2-胺母核上进行结构改造,例如在母核上并上一个哌啶环,得到了一类以5,6,7,8-四氢吡唑并[1,5-a]吡啶并[4,3-d]嘧啶-2-胺为母核的化合物得到代表化合物35,和改变并环上N原子的位置排布得到以咪唑并[1,2-b]哒嗪-2-胺为母核的代表化合物36,化合物35和36对ATR均具有良好的Ki值,更多的活性数据有待公开。
7. 其他类
诺华公司通过高通量技术发现了先导化合物37 (见图11),对ATR的激酶IC50值为26 nM,但选择
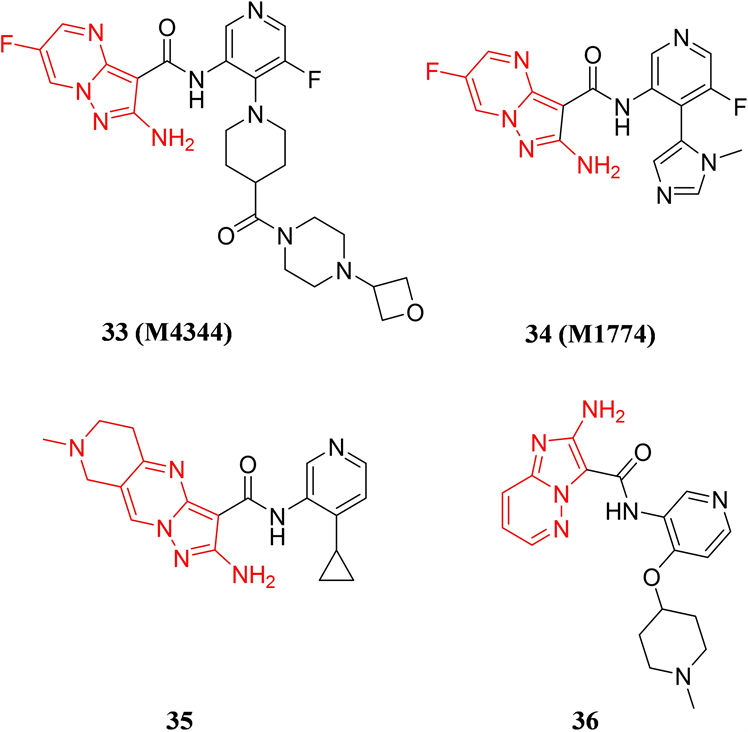
Figure 10. The chemical structures of compound 33-36
图10. 化合物33-36的化学结构
性较差,对PI3Kα的激酶IC50值为9 nM。通过对母核、伸向后口袋的双甲氧基苯基和伸向溶剂域的苯甲酰胺基团进行结构优化得到了以(R)-6-甲基-4,5,6,7-四氢吡唑并[1,5-a]吡嗪为核心的高活性及选择性的化合物38 (ATR IC50 = 0.4 nM, ATM/DNA-PK/mTOR IC50 = 4.3/1.6/4.4 μM),分析化合物38的结构类似物与ATR的结合模式,母核吡唑部分的N原子同铰链区产生关键性氢键相互作用,氮杂吲哚结构占据疏水区与并与区域附近的氨基酸残基形成双齿氢键。虽然化合物38的体外活性和选择性优越,但会对CYP3A4酶产生时间依赖性抑制,安全性问题限制了该化合物的进一步开发研究 [44] 。
先导化合物39 (见图12)同样由诺华公司通过高通量筛选发现,其对ATR具有中等程度抑制活性,并对PI3Kα具有良好的选择性(ATR IC50 = 96 nM, PI3Kα IC50 > 10 μM)。通过一系列的结构改造得到了1H-咪唑并[4,5-c]吡啶类化合物40,与化合物40相比,对ATR的激酶活性得到显著提升(ATR IC50 = 0.5 nM),对PIKK家族其他激酶成员也表现出很高的选择性。此外,化合物40的成药性良好(solubility > 2400 μM; hERG IC50 > 30 μM; CYP IC50: all > 24 μM),具有一定的临床研究价值。分析化合物40的结构类似物与蛋白的结合模式,吗啉结构中的N原子同铰链区氨基酸残基形成一处氢键,吲哚结构占据后口袋疏水区,与母核相连的环丙基甲基伸向蛋白的溶剂域 [45] 。

Figure 11. (A) The chemical structures of compound 37-38. (B) Molecular docking model of analogue of compound 38 with ATR [25]
图11. (A) 化合物37-38的化学结构;(B) 化合物38的结构类似物与ATR的结合模式

Figure 12. (A) The chemical structures of compound 39-40. (B) Molecular docking model of analogue of compound 40 with ATR [45]
图12. (A)化合物39-40的化学结构;(B)化合物40的结构类似物与ATR的结合模式
麦迪韦逊医疗公司对内BTK和PI3Kδ双靶抑制剂化合物库进行激酶筛选,得到对ATR具有中等抑制活性,且仅对BTK和PI3Kδ产生微弱抑制的先导化合物41 (ATR IC50 = 0.212 μM) (见图13)。通过结构优化进一步得到了激酶活性提升3倍的化合物43 (ATR IC50 = 0.066 μM)。化合物42对ATR具有一定的选择性,对ATM和DNA-PK的抑制活性较弱(ATM:抑制率 = 28% at 1 μM;DNA-PK:抑制率 = 59% at 1 μM)。但化合物在小鼠体内的口服生物利用度较低,仅为2.83%,提示仍需对其进行进一步的成药性改造。
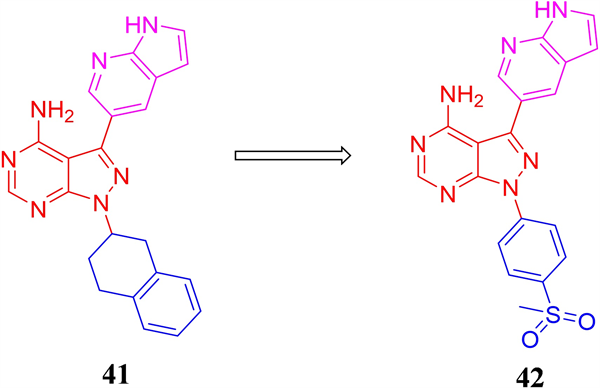
Figure 13. The chemical structures of compound 41-42
图13. 化合物41-42的化学结构
微芯药业公开了一类吡嗪酮类化合物作为ATR抑制剂的专利,代表化合物43 (见图14)对ATR激酶抑制IC50值为0.015 μM。该类化合物的设计思路推测源自AZD6738,将嘧啶核心替换为吡嗪酮结构后,再对其他片段进行优化 [46] 。

Figure 14. The chemical structure of compound 43
图14. 化合物43的化学结构
4. 结语
ATR作为DNA损伤反应中的一个关键蛋白,对肿瘤的存活至关重要,这使其成为一个很有吸引力的抗癌靶点。靶向ATR是许多抗肿瘤药物的研究重点,目前已报道了多种ATR抑制剂,按结构可分为吡嗪类、嘧啶类、吡啶类、稠环嘧啶类、稠环吡啶类和其他类等,其中有10多种小分子抑制处于不同临床研究阶段。尽管在这一领域取得了令人鼓舞的进展,但还存在缺乏选择性和没有合适的生物标志物来指导潜在的用药组合等问题。确定和选择合适的患者组进行单药治疗,以及选择适当的剂量和合理安排ATR抑制剂进行联合治疗依然是今后研究的重点。
文章引用
程浩东,段云鑫,施志浩. ATR小分子抑制剂的研究进展
Advances in the Study of Small Molecule Inhibitors of ATR[J]. 药物资讯, 2023, 12(02): 138-156. https://doi.org/10.12677/PI.2023.122018
参考文献
- 1. Choi, W.Y. and Lee, E.S. (2022) Therapeutic Targeting of DNA Damage Response in Cancer. International Journal of Molecular Sciences, 23, 1701. https://doi.org/10.3390/ijms23031701
- 2. Desai, A., Yan, Y. and Gerson, S.L. (2018) Advances in Therapeutic Targeting of the DNA Damage Response in Cancer. DNA Repair, 66-67, 24-29. https://doi.org/10.1016/j.dnarep.2018.04.004
- 3. Jackson, S.P. and Helleday, T. (2016) Drugging DNA Repair. Science, 352, 1178-1179. https://doi.org/10.1126/science.aab0958
- 4. Roos, W.P., Thomas, A.D. and Kaina, B. (2016) DNA Damage and the Balance between Survival and Death in Cancer Biology. Nature Reviews Cancer, 16, 20-33. https://doi.org/10.1038/nrc.2015.2
- 5. Poehlmann, A. and Roessner, A. (2010) Importance of DNA Damage Checkpoints in the Pathogenesis of Human Cancers. Pathology Research and Practice, 206, 591-601. https://doi.org/10.1016/j.prp.2010.06.006
- 6. Yazinski, S.A. and Zou, L. (2016) Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway. Annual Review of Genetics, 50, 155-173. https://doi.org/10.1146/annurev-genet-121415-121658
- 7. Cimprich, K.A. and Cortez, D. (2008) ATR: An Es-sential Regulator of Genome Integrity. Nature Reviews Molecular Cell Biology, 9, 616-627. https://doi.org/10.1038/nrm2450
- 8. Foote, K.M., Lau, A. and Nissink, J.W.M. (2015) Drugging ATR: Progress in the Development of Specific Inhibitors for the Treatment of Cancer. Future Medicinal Chemistry, 7, 873-891. https://doi.org/10.4155/fmc.15.33
- 9. Taylor, E.M. and Lindsay, H.D. (2016) DNA Replication Stress and Cancer: Cause or Cure? Future Oncology, 12, 221-237. https://doi.org/10.2217/fon.15.292
- 10. Lecona, E. and Fernan-dez-Capetillo, O. (2018) Targeting ATR in Cancer. Nature Reviews Cancer, 18, 586-595. https://doi.org/10.1038/s41568-018-0034-3
- 11. Cimprich, K.A., Shin, T.B., Keith, C.T., et al. (1996) cDNA Cloning and Gene Mapping of a Candidate Human Cell Cycle Checkpoint Protein. Proceedings of the National Academy of Sciences of the United States of America, 93, 2850-2855. https://doi.org/10.1073/pnas.93.7.2850
- 12. Dumaz, N. and Meek, D.W. (1999) Serine15 Phosphorylation Stimulates p53 Transactivation but Does Not Directly Influence In-teraction with HDM2. EMBO, 18, 7002-7010. https://doi.org/10.1093/emboj/18.24.7002
- 13. Halazonetis, T.D., Gorgoulis, V.G. and Bartek, J. (2008) An Oncogene-Induced DNA Damage Model for Cancer Development. Science, 319, 1352-1355. https://doi.org/10.1126/science.1140735
- 14. Hu, S., Hui, Z., Duan, J., Garrido, C., Xie, T. and Ye, X.Y. (2022) Discovery of Small-Molecule ATR Inhibitors for Potential Cancer Treatment: A Patent Review from 2014 to Present. Expert Opinion on Therapeutic Patents, 32, 401-421. https://doi.org/10.1080/13543776.2022.2027911
- 15. Ronald, K., Jean-Damien, C., Steven, D., Davis, C., O’Donnell, M., Storck, P., et al. (2019) Rational Design of 5-(4-(Isopropylsulfonyl)phenyl)-3-(3-(4-((methylamino)methyl)phenyl)isoxazol-5-yl)pyrazin-2-amine(VX-970, M6620): Optimization of Intra- and Intermolecular Polar Interactions of a New Ataxia Telangiectasia Mutated and Rad3-Related (ATR) Kinase Inhibitor. Journal of Medicinal Chemistry, 62, 5547-5561. https://doi.org/10.1021/acs.jmedchem.9b00426
- 16. Kevin, F., Willem, N., Thomas, M., et al. (2018) Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent. Journal of Medicinal Chemistry, 61, 9889-9907. https://doi.org/10.1021/acs.jmedchem.8b01187
- 17. Francesco, D., et al. (2019) Heterocyclic Inhibitors of ATR Kinase: WO2019014618.
- 18. Zhao, Y.P., Liu, B., Jiang, Y.Y., et al. (2021) 2,4,6-Tri-substituted Pyrimidine Com-pounds as ATR Kinase Inhibitors: WO2021233376. 2021-11-25.
- 19. Qian, W.Y., Wang, J., Li, J., Li, J., et al. (2019) ATR Inhibitor and Application Thereof: WO2019154365. 2019-08-15.
- 20. Francesco, D., et al. (2019) Heterocyclic Inhibitors of ATR Kinase: WO2019178590. 2019-09-19.
- 21. Shan, B., Hou, B., Yu, W.H., et al. (2022) ATR Inhibi-tors and Uses Thereof: WO2022002245. 2022-01-06.
- 22. Fang, K. and Chen, S.H. (2021) Fluoropyrrolopyridine Compound and Application Thereof: WO2021238999. 2021-12-02.
- 23. Bin, H.C., Chen, P., Wu, M., et al. (2022) Discovery of a Potent and Highly Selective Inhibitor of Ataxia Telangiectasia Mutated and Rad3-Related (ATR) Kinase: Structural Activity Relationship and Antitumor Activity both in Vitro and in Vivo. European Journal of Medicinal Chem-istry, 232, Article ID: 114187. https://doi.org/10.1016/j.ejmech.2022.114187
- 24. Chen, P., Bin, H.C., Jiao, Y., et al. (2022) Discovery of 6,7-Dihydro-5H-pyrrolo[3,4-d] Pyrimidine Derivatives as a New Class of ATR Inhibitors. Bioorganic & Medicinal Chemistry Letters, 63, Article ID: 128651. https://doi.org/10.1016/j.bmcl.2022.128651
- 25. Li, X., Chen, Y., Cai, G.D., et al. (2022) Imidazopyrimidine De-rivative, Preparation Method Therefor and Medical Use Thereof: WO2022002243. 2022-01-06.
- 26. 李心, 杨芳, 冯斌强, 等. 稠合嘧啶类衍生物、其制备方法及其在医药上的应用[P]. 中国, CN113135942. 2021-07-20.
- 27. Li, X. Zeng, C.G., He, F., et al. (2021) Fused Heteroaryl Derivative, Preparation Method Therefor, and Application Thereof in Medicine: WO2021143821. 2021-07-22.
- 28. Zhao, Z.M., Yang, W., Wu, Z.H., et al. (2020) Heterocyclic Fused Py-rimidine Derivative, Pharmaceutical Composition Thereof, and Application Thereof: WO2020103897. 2020-05-28.
- 29. Lücking, U., Wortmann, L., Wengner, A.M., et al. (2020) Damage Incorporated: Discovery of the Potent, Highly Selective, Orally Available ATR Inhibitor BAY 1895344 with Favorable Pharmacokinetic Properties and Promising Efficacy in Monotherapy and in Combination Treatments in Preclinical Tumor Models. Journal of Medicinal Chemistry, 63, 7293-7325. https://doi.org/10.1016/j.bmcl.2022.128651
- 30. Wengner, A.M., Siemeister, G., Lücking, U., et al. (2020) The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage-Inducing or Repair-Compromising Therapies in Preclinical Cancer Models. Molecular Cancer Therapeu-tics, 19, 26-38. https://doi.org/10.1158/1535-7163.MCT-19-0019
- 31. Yap, T.A., Tan, D.S.P., Terbuch, A., et al. (2021) First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Pa-tients with Advanced Solid Tumors. Cancer Discovery, 11, 80-91. https://doi.org/10.1158/2159-8290.CD-20-0868
- 32. 刘金明, 张浩亮, 何婷, 等. 一种取代吡啶并并[3,4-b]吡嗪-2(1H)-酮化合物、其制备方法和用途[P]. 中国, CN111848605. 2020-10-30.
- 33. Burgdorf, L. and Tsaklakidis, C. (2020) 5-Morpholin-4-yl-pyrazolo[4,3-b]pyridine derivatives: WO2020049017. 2020-03-12.
- 34. 吴颢, 沈益飞, 姚智理, 等. ATR抑制剂及其在医药上的应用[P]. 中国, CN112851668. 2021-05-28.
- 35. Li, X., Dong, H.D., Bai, D.D., et al. (2021) Pyrazolo-Heteroaryl Derivative, Preparation Method Therefor, and Medical Use Thereof: WO2021098811. 2021-05-27.
- 36. Shan, B., Hou, B., Yu, W.H., et al. (2022) Atr Inhibitors and Uses Thereof: WO2022028598. 2022-02-10.
- 37. Cai, S.X., Tian, Y. and Wang, X.Z. (2020) Substituted Fused Heteroaromatic Bicyclic Compounds as Kinase Inhibitors and the Use Thereof: WO2020259601. 2020-12-30.
- 38. Cheng, C., Zhao, Y.P., Wang, H.J., et al. (2022) Pyrazolopyrimidine Compound Used as Atr Kinase Inhibitor: WO2022012484. 2022-01-20.
- 39. Roulston, A., Zimmermann, M., Papp, R., et al. (2022) RP-3500: A Novel, Potent, and Selective ATR Inhibitor That Is Effective in Preclinical Models as a Monotherapy and in Combination with PARP Inhibitors. Molecular Cancer Therapeutics, 21, 245-256. https://doi.org/10.1158/1535-7163.MCT-21-0615
- 40. 刘晓辉, 刘凤涛, 高大新. 三杂环衍生物、其药物组合物及应用[P]. 中国, CN114369096. 2022-04-19.
- 41. Wang, S.H., Yang, C.D. and Chen, S.H. (2022) Class of 1,7-Naphthyridine Compounds: WO2022063308. 2022-03-31.
- 42. Jo, U., Senatorov, I.S., Zimmermann, A., et al. (2021) Novel and Highly Potent ATR Inhibitor M4344 Kills Cancer Cells with Replication Stress, and Enhances the Chemotherapeutic Activity of Widely Used DNA Damaging Agents. Molecular Cancer Therapeutics, 20, 1431-1441. https://doi.org/10.1158/1535-7163.MCT-20-1026
- 43. Yap, T.A., Tolcher, A.W., Plummer, E.R., et al. (2021) A First-in-Human Phase I Study of ATR Inhibitor M1774 in Patients with Solid Tumors. Journal of Clinical Oncology, 39, TPS3153. https://doi.org/10.1200/JCO.2021.39.15_suppl.TPS3153
- 44. Paul, B., Robert, A., Xianming J, et al. (2015) Structure-Based Drug Design of Novel Potent and Selective Tetrahydropyrazolo[1,5-a]pyrazines as ATR Inhibitors. ACS Medicinal Chemistry Letters, 6, 37-41. https://doi.org/10.1021/ml500353p
- 45. Paul B, Yue P, Yipin L, et al. (2015) Structure-Based Drug Design of Novel, Potent, and Selective Azabenzimidazoles (ABI) as ATR Inhibitors. ACS Medicinal Chemistry Letters, 6, 42-46. https://doi.org/10.1021/ml500352s
- 46. 王伟, 曾宏, 巩晓明, 等. 一类吡嗪酮衍生物、其制备及其应用[P]. 中国, CN114591324. 2022-06-07.