Hans Journal of Nanotechnology
Vol.
11
No.
03
(
2021
), Article ID:
44639
,
9
pages
10.12677/NAT.2021.113022
低自旋(S = 1/2)过渡金属配合物的慢磁弛豫
徐红娟,刘泉*
南通大学化学与化工学院,江苏 南通

收稿日期:2021年7月6日;录用日期:2021年8月8日;发布日期:2021年8月19日

摘要
随着单离子磁体的蓬勃发展,研究人员发现即使当配合物的基态自旋为低自旋(S = 1/2),仍可以表现慢磁弛豫行为。这类配合物在量子信息处理方面(QIP)方面具有较大的应用潜能,因此得到了众多研究者的关注。对于过渡金属配合物,配体的选择影响中心金属离子的电子结构,进而影响其慢磁弛豫行为及量子相干性质。本文结合近年来的研究成果,对低自旋配合物的慢磁弛豫行为进行了阐述,分析了其弛豫行为来源,并对弛豫机理进行总结,为量子比特的设计提供思路。
关键词
过渡金属,低自旋,慢磁弛豫,弛豫机理

Slow Magnetic Relaxation in Low-Spin (S = 1/2) Transition Metal Complexes
Hongjuan Xu, Quan Liu*
College of Chemistry and Chemical Engineering, Nantong University, Nantong Jiangsu
Received: Jul. 6th, 2021; accepted: Aug. 8th, 2021; published: Aug. 19th, 2021

ABSTRACT
With the rapid development of single ion magnets, researchers found that a few of complexes even with low-spin (S = 1/2) ground state spin can still exhibit slow magnetic relaxation behavior. Owing to their great potential in quantum information processing (QIP), these complexes have attracted many researchers’ attention. For transition metal complexes, the ligands coordinating to the transition metals have great impact on the electronic structure of central metal ions, and then significantly affect the slow magnetic relaxation behavior and quantum coherence properties. Based on the recent research, this paper describes the slow magnetic relaxation behavior in low-spin complexes. Moreover, we have analyzed the origin of the relaxation behavior and summarized the relaxation mechanism in these complexes, which will provide new routes in the design of quantum qubit.
Keywords:Transition Metal, Low-Spin, Slow Magnetic Relaxation, Relaxation Mechanism

Copyright © 2021 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/


1. 引言
分子磁体在近二十年来取得飞速发展并成为化学、物理、材料等多个领域的研究热点。自从在十二核锰络合物Mn12Ac [1] 中观测到来源于孤立分子的磁双稳态后,越来越多的具有不同金属离子的配合物被报道表现出了缓慢的磁弛豫行为,这类配合物被统称为单分子磁体(SMMs)。由于其结构和性质易于设计和调控,因此有望作为新一代多功能磁性材料广泛应用于信息技术 [2]、自旋电子器件 [3] 以及量子计算 [4] 等领域。对于SMMs,当其自旋基态S > 1/2时,对称性的降低会导致激发态的分裂,然后通过自旋轨道耦合与基态混合,从而产生较大的磁各向异性,并表现出经典的单分子磁体行为 [5]。
另一方面,当自旋基态S = 1/2时,体系中不存在与零场分裂相关的自旋翻转能垒 [5],因此很难观测到慢磁弛豫行为。但是研究发现,某些刚性钒铜配合物在高温下可表现出较长的磁弛豫时间 [6] [7] [8] [9]。而这些同时具有较小自旋值和慢磁弛豫行为的金属配合物可能在量子计算机的量子比特器件中发挥重要作用,因此吸引了众多研究人员的注意。对于经典计算,数据以二进制数字编码,每个二进制数字都是一个定义良好的状态(0或1)。相比之下,量子比特则是任意量子态的叠加 [10] [11] [12]。自旋值较小的磁性分子如果能达到较长的退相干时间,便可成为一个好的量子位,甚至可用于建立密集的二维量子比特网络。迄今为止,在众多不同的配合物 [13] [14] 中均发现了适合量子比特的物理载流子。其中,基于特殊单分子磁体,即单离子磁体(SIMs),的体系成为了最近的研究热点。这是由于其化学合成和磁构关系较为简单,可以系统地阐明影响量子退相干的主要因素,包括自旋的大小和自旋轨道耦合效应以及其他外围因素,如配位配体等,从而可以更好地指导量子比特的合成设计。
为了实现量子干涉,提高相干值,应尽可能选择磁各向异性较小的低自旋配合物,比如说具有较弱自旋–轨道贡献的过渡金属配合物,而不是较重的稀土金属配合物。在这种S = 1/2的低自旋体系中,由于缺乏由热激发引起的中间电子态 [15],所以其慢磁弛豫不包含奥巴赫过程,而大都为拉曼过程。本文系统总结了近年来报道的不同的低自旋过渡金属配合物,并针对其在不同配位环境中表现出的慢磁弛豫行为等进行了阐述。
2. 低自旋过渡金属配合物
目前报道的具有慢磁弛豫行为的低自旋(S = 1/2)过渡金属离子主要包括V(IV) [6] [7] [16],Mn(IV) [17],Fe(III) [18],Co(II) [19] [20],Ni(I/III) [21] [22] [23],Cu(II) [24],Ru(III) [25] 和Ir(IV) [26]。
2.1. 低自旋V(IV)配合物
Sessoli课题组报道了一系列具有慢磁弛豫行为的V(IV)配合物,且全部为五配位和六配位 [6] [7] [16]。其中,2016年该课题组报道了一例五配位化合物VOPc [7]。中心V(IV)离子与四个N原子和一个O原子形成了畸变四方锥构型(图1)。交流磁化率表明当温度在20 K以内时,该配合物在高场0.2 T和1.0 T下均表现慢磁弛豫行为,而且弛豫的主要过程为直接过程。当VOPc与同构抗磁性主体TiOPc以1:1000的摩尔比混合时,在室温下可以检测到量子相干性。这种基于电子自旋的量子态可以有效地初始化,具有很高的热稳定性,是一种很有应用潜能的量子比特。

Figure 1. Molecular structure of VOPc
图1. VOPc的分子结构图
2.2. 低自旋Mn(IV)配合物
2016年,Ding等人报道了一例低自旋四配位Mn(IV)单离子磁体PhB(MesIm)3Mn≡ = phenyltris (3-mesitylimidazol-2-ylidene)borato) [17]。Jahn-Teller效应导致该配合物呈畸变四面体构型(图2)。交流磁化率数据表明该配合物在外加场下表现出慢磁弛豫行为,为场致单离子磁体。通过EPR数据分析与理论计算证明该配合物的慢磁弛豫行为来源于自旋轨道耦合导致的磁各向异性。通过对不同外加场以及不同温度下弛豫时间的分析发现该配合物中的弛豫过程主要为量子隧穿和拉曼过程。

Figure 2. Molecular structure of PhB (MesIm)3Mn≡N
图2. PhB (MesIm)3Mn≡N的分子结构图
2.3. 低自旋Fe(III)配合物
最近Buades等人利用碳硼烷合成了第一例具有慢磁弛豫行为的低自旋(S = 1/2)Fe(III)化合物[NMe4] [3,30-Fe(1,2-C2B9H11)2] [18] (图3)。对该配合物的溶液进行交流磁化率测试发现,在零场下没有虚部交流磁化率信号,当施加0.05 T外场时,配合物表现出慢磁弛豫行为,主要弛豫过程为拉曼过程。理论计算表明虽然该配合物的基态自旋等于1/2,但是依然中存在自旋轨道耦合,由此产生了较大的磁各向异性,进而导致配合物出现慢磁弛豫现象。
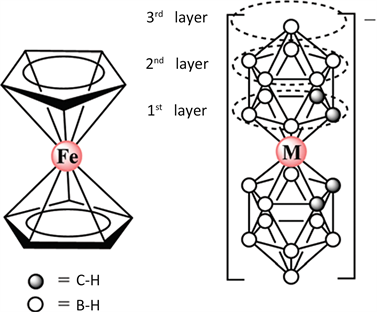
Figure 3. Molecular structure of [NMe4] [3,30-Fe(1,2-C2B9H11)2]
图3. [NMe4] [3,30-Fe(1,2-C2B9H11)2]的分子结构图
2.4. 低自旋Co(II)配合物
2015年,陈学太课题组利用1,4,7,10-四甲基-1,4,7,10-四氮杂环十二烷(12-TMC)以及两种不同的抗衡阴离子合成了两个具有四方单锥结构的五配位单核Co(II)配合物[Co(12-TMC)(CH3CN)](BF4)2和[Co(12-TMC)(CH3CN)](PF6)2 [19]。直流磁化率数据表明两个配合物中存在不完全自旋交叉现象(图4)。另外,交流磁化率数据显示两个配合物在外加场下均可表现出慢磁弛豫行为,且该行为来源于Co(II)离子的低自旋态。通过对弛豫时间的分析得出两个配合物中的弛豫过程主要为拉曼过程和直接过程。
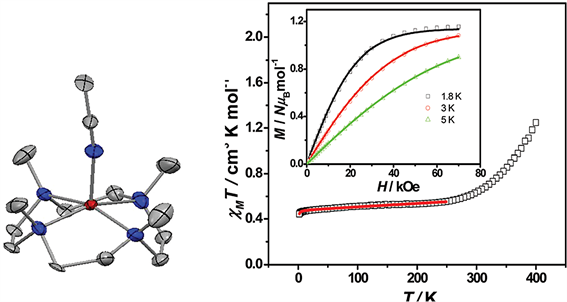
Figure 4. Molecular structure, variable-temperature susceptibility and magnetization of [Co(12-TMC)(CH3CN)](BF4)2
图4. [Co(12-TMC)(CH3CN)](BF4)2的分子结构、变温磁化率以及磁化强度图
此外,陈磊等人在2018年也报道了一例具有自旋交叉性质的五配位Co(II)配合物[Co(3,4-lut)4Br]Br (3,4-lut = 3,4-lutidine) [20]。该配合物的中心Co(II)离子在赤道位置上与四个N原子配位,轴向位置上与Br−离子配位,形成了扭曲四方锥构型(图5)。单晶结构衍射数据表明该配合物结构在常温和低温下存在微小差别。随着温度的降低,其中N-Co-N夹角增大而Br-Co-N夹角减小。直流磁化率数据表明配合物中存在完全自旋交叉行为。该配合物在外加场下具有慢磁弛豫,且弛豫过程主要为量子隧穿和拉曼过程。
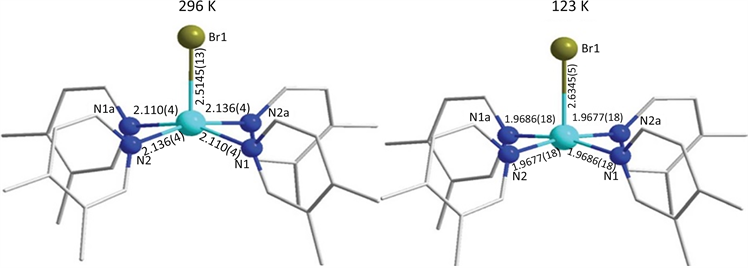
Figure 5. Molecular structure of [Co(3,4-lut)4Br]Br at different temperatures
图5. [Co(3,4-lut)4Br]Br在不同温度下的分子结构图
2.5. 低自旋Ni(I/III)配合物
低自旋镍配合物中的镍离子包括Ni(I)和Ni(III)两种价态。其中Ni(I)单离子磁体有两例,其慢磁弛豫行为的起因并不相同。第一例低自旋配合物是由Poulten等人在2013年报道的Ni(I)单离子磁体[Ni(6-Mes)2]Br (6-Mes = 1,3-bis(2,4,6-trimethylphenyl)-3,4,5,6-tetrahydropyrimidin-2-ylidene) [21]。该配合物为二配位,具有线型构型,C-Ni-C角为179.27(13)˚ (图6)。直流磁化率数据、理论计算和EPR谱图均证明该配合物中存在未猝灭的一级轨道角动量贡献。虽然该化合物中Ni(I)离子自旋态为S = 1/2,但是由一级旋轨耦合产生的较大磁各向异性使得该配合物在外加场下表现出慢磁弛豫行为。利用Arrhenius定律对不同温度下弛豫时间进行拟合,得到其能垒为11.8 cm−1。
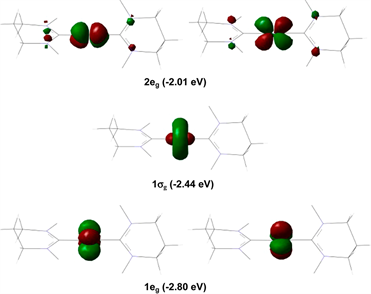
Figure 6. Molecular structure and orbital distribution of [Ni(6-Mes)2]+
图6. [Ni(6-Mes)2]+的结构图及轨道分布图
2016年,Lin等人报道了两例三配位Ni(I)配合物[NiCl(PPh3)2]∙C4H8O和[Ni(N(SiMe3)2)(PPh3)2] (图7) [22]。交流磁化率数据表明两个配合物在低温下均具有场致单离子磁体性质。与Poulten等人报道的二配位线型Ni(I)单离子磁体不同,配合物[NiCl(PPh3)2]·C4H8O和[Ni(N(SiMe3)2)(PPh3)2]的慢磁弛豫行为并不是由一级旋轨耦合导致的磁各向异性所引发的,而是由二级旋轨耦合导致的g因子各向异性引起的。

Figure 7. Molecular structure of [Ni(N(SiMe3)2)(PPh3)2]
图7. [Ni(N(SiMe3)2)(PPh3)2]的分子结构图
2018年,Bhowmick等人报道了一系列低自旋Ni(III)配合物[Ni(cyclam)(NO3)2](NO3),[Ni(cyclam)(NO3)2] (ClO4)和[Ni(cyclam)(NCS)2](ClO4) (cyclam = 1,4,8,11-tetraazacyclotetradecane) [23]。三个配合物均为六配位呈畸变八面体构型(图8),其中前两个配合物的中心Ni(III)离子分别与四个N原子和两个O原子配位,而第三个配合物的中心Ni(III)离子与六个N原子配位。不同的配位环境导致配合物的动态磁化学数据也有所不同。交流磁化率表明由O原子参与配位的两个配合物在外加场下均表现慢磁弛豫行为,相反完全由N原子配位
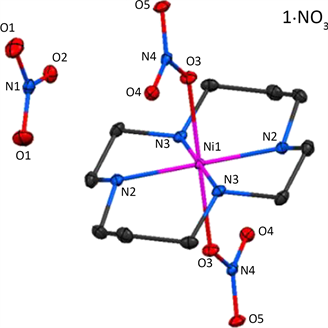
Figure 8. Molecular structure of [Ni(cyclam)(NO3)2](NO3)
图8. [Ni(cyclam)(NO3)2](NO3)的分子结构图
的配合物即使在外加场下依旧没有观测到慢磁弛豫现象。实验和理论计算均表明配合物中电子结构、分子内和分子间相互作用以及自旋和核自旋之间的耦合方式都会对慢磁弛豫行为产生影响。配合物[Ni(cyclam) (NO3)2](NO3)和[Ni(cyclam)(NO3)2](ClO4)中的弛豫过程主要包含拉曼过程和直接过程。
2.6. 低自旋Cu(II)配合物
Boča等人在2017年报道了一例六配位Cu(II)单离子磁体[Cu(pydca)(dmpy)]·0.5H2O (pydca = pyridine-2,6-dicarboxylato, dmpy = 2,6-dimethanolpyridine) [24],其结构如图9所示。交流磁化率数据表明该配合物在较大的外加场下表现出慢磁弛豫行为且具有双弛豫过程。其弛豫过程包含量子隧穿和拉曼过程。理论计算证明该配合物中存在由旋轨耦合导致的g因子各向异性,进而引发慢磁弛豫行为。
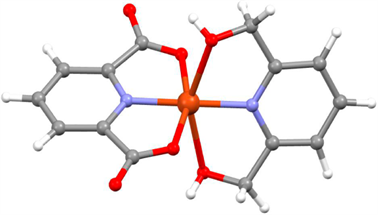
Figure 9. Molecular structure of [Cu(pydca)(dmpy)]·0.5H2O
图9. [Cu(pydca)(dmpy)]·0.5H2O的分子结构图
2.7. 低自旋Ru(III)和Ir(IV)配合物
研究人员不仅在3 d过渡金属低自旋配合物中观察到慢磁弛豫现象,而且在4 d和5 d过渡金属低自旋配合物中也观测到慢磁弛豫现象。
2017年,Wu等人报道了一例Ru(III)配合物[RuCl3(PPh3)2(MeCN)] (PPh3 = triphenylphosphine) [25]。如图10所示,该配合物为六配位,中心Ru(III)离子分别与三个Cl原子、两个P原子和一个N原子配位,

Figure 10. Molecular structure and electronic configuration of [RuCl3(PPh3)2(MeCN)]
图10. [RuCl3(PPh3)2(MeCN)]的分子结构及电子轨道分布图
呈八面体构型。交流磁化率表明该配合物在外加场下具有慢磁弛豫行为,且弛豫主要由声子瓶颈效应导致的。该研究表明配合物分子中的超精细作用可以捕获长波声子,从而减慢弛豫速率。
2016年,Pedersen等人报道了三个六配位Ir(IV)配合物(PPh4)2[IrF6]∙2H2O、(Zn(viz)4)[IrF6]和(PPh4)2[IrCl6] [26]。三个配合物中的Ir(IV)离子分别不同的卤素离子配位,配位构型均为畸变八面体(图11)。交流磁化率表明三个配合物在外加场下均具有慢磁弛豫行为,其中配合物(PPh4)2[IrF6]∙2H2O和Zn(viz)4[IrF6]的弛豫过程主要为由声子瓶颈效应导致的直接过程。
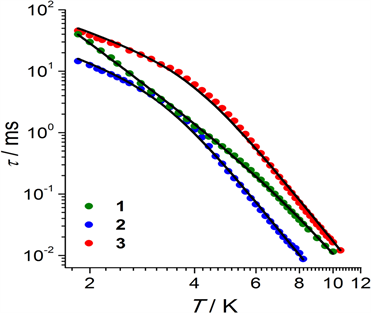
Figure 11. Molecular structureand relaxation time of six-coordinate Ir(IV) complexes
图11. 六配位Ir(IV)配合物的分子结构及弛豫时间
3. 结论与展望
理论上,基态S= 1/2的低自旋配合物很难表现出单分子磁体行为,但是越来越多的研究表明,在某些低自旋金属配合物中依旧可以观察到慢磁弛豫现象。这类配合物在量子计算方面有较大的应用潜能,因此在最近几年,基于单核过渡金属配合物的弛豫研究迅速发展。其中报道得最多的是单核3d过渡金属的低自旋配合物。随着研究的广泛和深入,逐渐出现了很多新的问题和挑战。首先,这类低自旋配合物虽然可以表现出慢磁弛豫,但是其弛豫来源和机理有待进一步的研究。但是随着各类表征手段的发展,对弛豫机理可能会有更加深刻的分析。其次,配体的选择,特别是与金属离子直接配位的原子,对中心金属离子的电子结构有很大影响,进而影响体系的慢磁弛豫行为。除此之外,溶剂和抗磁类配合物的浓度等也会对弛豫产生一定的影响。对这类因素的探究,亟需更多化学层面上的实验设计来证明。
基金项目
感谢国家自然科学基金(21776140)和南通科技项目基金(JCZ21022, JC2020134, JC2020133)的支持。
文章引用
徐红娟,刘 泉. 低自旋(S = 1/2)过渡金属配合物的慢磁弛豫
Slow Magnetic Relaxation in Low-Spin (S = 1/2) Transition Metal Complexes[J]. 纳米技术, 2021, 11(03): 191-199. https://doi.org/10.12677/NAT.2021.113022
参考文献
- 1. Sessoli, R., Gatteschi, D., Caneschi, A., et al. (1993) Magnetic Bistability in a Metal-Ion Cluster. Nature, 365, 141-143. https://doi.org/10.1038/365141a0
- 2. Leuenberger, M.N. and Loss, D. (2001) Quantum Computing in Molecular Magnets. Nature, 410, 789-793. https://doi.org/10.1038/35071024
- 3. Bogani, L. and Wernsdorfer, W. (2008) Molecular Spintronics Using Sin-gle-Molecule Magnets. Nature Materials, 7, 179-186. https://doi.org/10.1038/nmat2133
- 4. Wernsdorfer, W. and Sessoli, R. (1999) Quantum Phase Interference and Parity Effects in Magnetic Molecular Clusters. Science, 284, 133-135. https://doi.org/10.1126/science.284.5411.133
- 5. Ribas, J. (2008) Coordination Chemistry. Wiley-VCH, Wein-heim.
- 6. Atzori, M., Morra, E., Tesi, L., et al. (2016) Quantum Coherence Times Enhancement in Vanadi-um(IV)-Based Potential Molecular Qubits: The Key Role of the Vanadyl Moiety. Journal of the American Chemical So-ciety, 138, 11234-11244. https://doi.org/10.1021/jacs.6b05574
- 7. Atzori, M., Tesi, L., Morra, E., et al. (2016) Room-Temperature Quantum Coherence and Rabi Oscillations in Vanadyl Phthalocyanine: Toward Multifunctional Mo-lecular Spin Qubits. Journal of the American Chemical Society, 138, 2154-2157. https://doi.org/10.1021/jacs.5b13408
- 8. Graham, M.J., Zadrozny, J.M., Shiddiq, M., et al. (2014) Influence of Electronic Spin and Spin-Orbit Coupling on Decoherence in Mononuclear Transition Metal Complexes. Journal of the American Chemical Society, 136, 7623-7626. https://doi.org/10.1021/ja5037397
- 9. Bader, K., Dengler, D., Lenz, S., et al. (2014) Room Temperature Quantum Coherence in a Potential Molecular Qubit. Nature Communications, 5, 5304. https://doi.org/10.1038/ncomms6304
- 10. Bennett, C.H. and DiVincenzo, D.P. (2000) Quantum Information and Computation. Nature, 404, 247-255. https://doi.org/10.1038/35005001
- 11. Nielsen, M.A. and Chuang, I.L. (2010) Quantum Computation and Quan-tum Information. Cambridge University Press, Cambridge.
- 12. Aromi, G., Aguila, D., Gamez, P., et al. (2012) Design of Magnetic Coordination Complexes for Quantum Computing. Chemical Society Reviews, 41, 537-546. https://doi.org/10.1039/C1CS15115K
- 13. Sato, K., Nakazawa, S., Rahimi, R., et al. (2019) Molecular Elec-tron-Spin Quantum Computers and Quantum Information Processing: Pulse-Based Electron Magnetic Resonance Spin Technology Applied to Matter Spin-Qubits. Journal of Materials Chemistry, 19, 3739-3754. https://doi.org/10.1039/b819556k
- 14. Lehmann, J., Gaita-Ari Nmacr, A., Coronado, E., et al. (2007) Spin Qubits with Electrically Gated Polyoxometalate Molecules. Nature Nanotechnology, 2, 312-317. https://doi.org/10.1038/nnano.2007.110
- 15. Shrivastava, K.N. (1983) Theory of Spin-Lattice Relaxation. Physica Status Solidi B, 117, 437-458. https://doi.org/10.1002/pssb.2221170202
- 16. Tesi, L., Lucaccini, E., Cimatti, I., et al. (2016) Quantum Coherence in a Process Able Vanadyl Complex: New Tools for the Search of Molecular Spin Qubits. Chemical Science, 7, 2074-2083. https://doi.org/10.1039/C5SC04295J
- 17. Ding, M., Cutsail III, G.E., Aravena, D., et al. (2016) A Low Spin Manganese(IV) Nitride Single Molecule Magnet. Chemical Science, 7, 6132-6140. https://doi.org/10.1039/C6SC01469K
- 18. Buades, A.B., Arderiu, V.S., Maxwell, L., et al. (2019) Slow-Spin Re-laxation of a Low-Spin S = 1/2 FeIII Carborane Complex. Chemical Communications, 55, 3825-3828. https://doi.org/10.1039/C9CC01123D
- 19. Cui, H.H., Wang, J., Chen, X.T., et al. (2017) Slow Magnetic Relaxa-tion in Five-Coordinate Spin-Crossover Cobalt(II) Complexes. Chemical Communications, 53, 9304-9307. https://doi.org/10.1039/C7CC04785A
- 20. Chen, L., Song, J., Zhao, W., et al. (2018) A Mononuclear Five-Coordinate Co(II) Single Molecule Magnet with a Spin Crossover between the S = 1/2 and 3/2 States. Dalton Transactions, 47, 16596-16602. https://doi.org/10.1039/C8DT03783C
- 21. Poulten, R.C., Page, M.J., Algarra, A.G., et al. (2013) Synthesis, Elec-tronic Structure, and Magnetism of [Ni(6-Mes)2]+: A Two-Coordinate Nickel(I) Complex Stabilized by Bulky N-Heterocyclic Carbenes. Journal of the American Chemical Society, 135, 13640-13643. https://doi.org/10.1021/ja407004y
- 22. Lin, W., Bodenstein, T., Mereacre, V., et al. (2016) Field-Induced Slow Magnetic Relaxation in the Ni(I) Complexes [NiCl(PPh3)2]•C4H8O and [Ni(N(SiMe3)2)(PPh3)2]. Inorganic Chemistry, 55, 2091. https://doi.org/10.1021/acs.inorgchem.5b02497
- 23. Bhowmick, I., Roehl, A.J., Neilson, J.R., et al. (2018) Slow Magnetic Relaxation in Octahedral Low-Spin Ni(III) Complexes. Chemical Science, 9, 6564-6571. https://doi.org/10.1039/C7SC04482H
- 24. Boča, R., Rajnák, C., Titiš, J., et al. (2017) Field Supported Slow Mag-netic Relaxation in a Mononuclear Cu(II) Complex. Inorganic Chemistry, 56, 1478-1482. https://doi.org/10.1021/acs.inorgchem.6b02535
- 25. Wu, S.Q., Miyazaki, Y., Nakano, M., et al. (2017) Slow Magnetic Relaxation in a Mononuclear Ruthenium(III) Complex. Chemistry—A European Journal, 23, 10028-10033. https://doi.org/10.1002/chem.201702047
- 26. Pedersen, K.S., Bendix, J., Tressaud, A., et al. (2016) Iridates from the Molecular Side. Nature Communications, 7, Article No. 12195. https://doi.org/10.1038/ncomms12686