Material Sciences
Vol.07 No.01(2017), Article ID:19395,11
pages
10.12677/MS.2017.71002
Investigation of Electronic Structures of SrTiO3(001) Surface with N Adsorption by First-Principles
Limin Fang1, Kelun Zhao2,3*
1Department of Physics, Guangdong University of Education, Guangzhou Guangdong
2School of Mechanical and Automotive Engineering, South China University of Technology, Guangzhou Guangdong
3Zhuhai ROSSINI Watch Industry Ltd., Zhuhai Guangdong

Received: Nov. 30th, 2016; accepted: Dec. 24th, 2016; published: Dec. 27th, 2016
Copyright © 2017 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



ABSTRACT
It is well known that N doped SrTiO3 has been successfully used to develop semiconductor diodes in a blue-light region, transparent conductive films and perovskite-oxides hetero-junction optical- electric devices. The first-principles Plane-Wave PseudoPotential (PWPP) calculations have been performed to investigate the atomic configuration, chemical bond characteristics, and electronic structure of N atomic adsorption on SrTiO3(001) surface with the explanations of the related experiment phenomena in the present study. These results indicated that the stability of the adsorbed N on the surface depends on the relative position of N atom to the surface by analysis of adsorption energy, substitution energy and density of states.
Keywords:Adsorption, Surface, Electronic Structure, Atomic Configuration, Chemical Bond Characteristics, First-Principles

N吸附SrTiO3(001)表面电子结构的第一性原理研究
房丽敏1,赵可沦2,3*
1广东省第二师范学院物理系,广东 广州
2华南理工大学机械与汽车工程学院,广东 广州
3珠海罗西尼表业有限公司,广东 珠海

收稿日期:2016年11月30日;录用日期:2016年12月24日;发布日期:2016年12月27日

摘 要
N掺杂SrTiO3材料已成功用于研制蓝光波段发光二极管、高透明导电薄膜和钙钛矿氧化物光电子器件。本文采用第一性原理平面波赝势方法对N吸附SrTiO3(001)表面的原子构型、化学键特征和电子结构进行了计算研究并解释了In掺杂SrTiO3导电薄膜的相关实验现象。通过吸附能、置换能和态密度的比较分析发现,SrTiO3(001)表面吸附N原子的稳定性与N原子相对表面层的位置密切相关。
关键词 :吸附,表面,电子结构,原子构型,化学键特征,第一性原理

1. 引言
在众多氧化物催化剂中,钙钛矿型SrTiO3具有耐光化学腐蚀好,性能稳定,禁带宽度较为适合和导带电位比金红石型TiO2更低的特点,因此SrTiO3是一种具有极大应用前景的光催化材料 [1] 。纯SrTiO3本身具有一定的光催化活性,但其较大的光学带隙(3.25 eV)限制了其实际应用;为了提高纯SrTiO3的光催化活性,人们参考掺杂TiO2系列催化材料的方法探索各种SrTiO3掺杂物的光催化功效以及微观机制。因此,N掺杂SrTiO3材料光催化性质和电子结构的微观机制引起了人们的广泛研究 [2] [3] [4] [5] 。首先,Wang等 [2] 运用机械化学法制备了单相N掺杂SrTiO3光催化剂,为制备单相掺杂SrTiO3光催化剂提供了一种简单而有效的方法。Mi等 [3] 利用X射线光电子发射谱和第一性原理方法分析了N掺杂效应对SrTiO3 薄膜电子结构和光学性质的影响,位于O-2p价带边上方的局域N-2p价带电子态导致新的带隙形成。Liu等 [4] 运用离子注入技术制备了N掺杂SrTiO3单晶材料,分析了N掺杂前后的样品晶格属性与光学性质。Marozau等 [5] 运用PRCLA和RFPLD两种方法,分别制备了沉积在SrTiO3(001),LaAlO3(001)和MgO(001)衬底表面的N掺杂SrTiO3薄膜;研究发现,利用PRCLA方法生成的N-SrTiO3薄膜中N原子的掺杂浓度高于RFPLD方法,且N原子掺杂浓度可通过调整沉积参数进行适当调节。由于大多数光催化性能的材料涉及到表面处理,而且光催化材料的表面掺杂可能会影响某些材料表面的某些性质。例如,对SrTiO3表面相关性质的研究发现,表面缺陷 [6] [7] [8] [9] 以及表面吸附效应 [10] [11] 将直接导致其表面原子结构畸变和电学性能以及化学反应微观机制的变化,因此分析N吸附SrTiO3表面的原子结构变化与电子结构特征具有重要的实际意义。所谓第一性原理平面波赝势方法,就是在求解单电子薛定锷方程时,用平面波展开电子的波函数ψn(k, r),对有效势VH(r)采取赝势近似。第一性原理平面波赝势方法目前已经成为固体能带计算中最成熟、应用最广泛的方法之一,并且已成功用于研究SrTiO3表面/界面材料的原子结构和化学吸附现象 [12] [13] 。本文采用第一性原理平面波赝势方法,讨论分析了N原子在SrTiO3(001)表面不同位置的吸附能和置换能以及吸附N原子的稳定性,并系统研究了N原子吸附SrTiO3(001)表面的原子构型及其化学键特征的变化。
2. 实验
本文采用第一性原理方法来研究N吸附SrTiO3(001)表面的结构特征和电子结构。理论计算程序采用基于密度泛函理论平面波赝势(PWPP)方法的CASTEP程序包 [14] [15] 。交换关联相互作用采用CA-PZ参数化形式的局域密度近似(LDA) [16] [17] 方法来处理,而对于Sr、Ti、O、N等原子的赝势则采用Vanderbilt形式的超软赝势。在计算过程中,平面波的截止能量设为350 eV (25.7 Ry),它反映了基于平面波基矢各计算特性的收敛度。对于整个布里渊区的积分计算采用Monkhorst-Pack形式的特殊k点方法,选取k点的数目也可以经过优化,以保证系统能量收敛。通过共轭梯度法使基于平面波基矢的体系总能达到最小值。自洽场(SCF)计算采用基于Pulay算法的混合密度方案。SCF的容许误差设定为1 × 10−6 eV/atom。
为了计算研究N原子在SrTiO3(001)表面的吸附效应,我们建立了图1所示三维周期性的SrTiO3(001)表面薄层超胞模型。图1(a)是以TiO2原子层截止的1 × 1 SrTiO3(001)表面薄层超胞示意图,其中包括4个TiO2以及3个SrO原子层,共含有18个原子;图1(b)是以SrO原子层截止的1 × 1 SrTiO3(001)表面薄层超胞,其中共包括4个SrO以及3个TiO2原子层,共含有17个原子。二者都是选用了包含7个原子层的薄层结构并含有两层对称且厚度为11 Å的真空层,这足以满足计算结果收敛性并避免顶部和底部的相互作用 [16] [17] [18] ,并且此模型具有以中间层为镜面的周期对称性。其中,I, II, III (1, 2, 3)分别代表薄层超胞内(图1(a)和图1((b)所示)的表面层、次表面层和第三原子层。在此基础上,我们建立了2 × 2 SrTiO3(001)表面薄层模型来分析N原子吸附后SrTiO3(001)表面结构特征和电学性质的变化规律,其中包括N直接吸附在表面层上和N置换表面薄层中O原子两种情况。对于N原子直接吸附的表面模型,图1(c)中逐个标明了对应N原子在表面层的不同吸附位置,即表面层原子Tis(I), Os(I), Srs(1)和Os(1)以及间隙位置A和B。对于N原子置换吸附表面模型,N原子分别置换位于表面层、次表面层和第三原子层上的Os(I)、Osub(II)和Ord(III)(Os(1), Osub(2), Ord(3))原子。在对SrTiO3(001)表面薄层超胞模型的几何优化过程中,所有结构参数都不受空间群对称性的限制;因此,经过连续迭代过程,就可以使超胞几何结构优化以获得一个稳定的结构;从而调节原子坐标和一些可能的超胞参数降低结构总能。
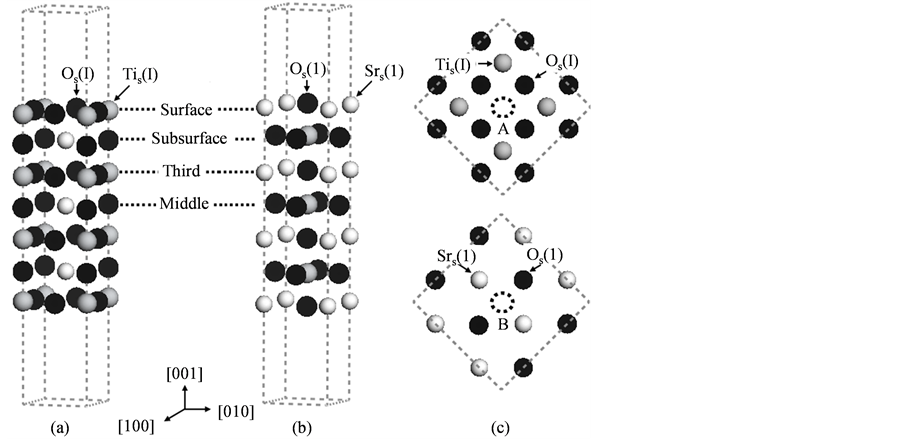
Figure 1. Schematics of the computational models of the SrTiO3(001) surface in present study. The 1 × 1 supercells of the slab model with (a) TiO2-terminated and (b) SrO-terminated SrTiO3(001) surface, respectively; (c) Top view of the surface atomic layer of the 2 × 2 supercells, in which we illustrate the different adsorption sites for N on the surface layer
图1. 本文理论计算的表面模型示意图。(a)和(b)分别代表TiO2和SrO原子层截止SrTiO3(001)表面的1 × 1薄层超胞模型;(c)则是2 × 2表面薄层超胞模型的俯视图,其中标明了N原子在表面层的不同吸附位置
3. 结果与讨论
3.1. 表面原子结构
为了比较SrTiO3(001)表面上不同吸附位置N原子的相对稳定性,在每个表面模型结构优化完成后,分别求出相应N原子的吸附能和置换能,并仔细分析N原子吸附效应对SrTiO3(001)表面原子结构的影响。
3.1.1. N原子直接吸附SrTiO3(001)表面
对于N直接吸附的表面模型,N原子的吸附能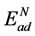 可以表示为:
可以表示为:
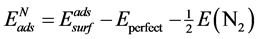 (1)
(1)
式中: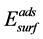 和
和 分别表示N吸附表面模型和理想表面模型结构优化的总能。E(N2)则代表一个孤立N2分子的自旋极化总能 [19] 。为了进行比较,我们也计算了一个孤立N原子的吸附能,即在公式(1)中用E(N)代替1/2E(N2),而E(N)则代表一个孤立N原子(3p电子态)的自旋极化总能 [20] 。根据公式(1)得到的吸附能若为负值则表示SrTiO3(001)表面和N原子之间相互吸引,因此由公式(1)定义的吸附能反映了表面不同位置对N原子的吸附强度。我们在表1以及相应的讨论过程中采用了吸附能的绝对值,这也意味着N原子吸附能量值越大则相应表面位置吸附的N原子更为稳定。据我们所知:DFT-LDA方法易于高估材料的结合能(Binding energy),而结合能的精确度主要取决于交换-关联泛函(exchange-correlation functional)的选取形式即不同的交换-关联泛函都会导致其计算结果与结合能绝对值(实验值)存在一定的差异。关于SrTiO3表面吸附的理论研究 [18] 发现:交换关联泛函即使是分别选取LDA近似和GGA近似,相应结合能计算结果之间仅仅存在定性的差异,而这两种方法的数值结果具有相同的变化趋势和结论。本文工作主要考虑N原子吸附能和置换能的相对变化趋势而不是其绝对值的大小,因此本文研究的主要结论不会因为选择算法的差异而受到干扰。
分别表示N吸附表面模型和理想表面模型结构优化的总能。E(N2)则代表一个孤立N2分子的自旋极化总能 [19] 。为了进行比较,我们也计算了一个孤立N原子的吸附能,即在公式(1)中用E(N)代替1/2E(N2),而E(N)则代表一个孤立N原子(3p电子态)的自旋极化总能 [20] 。根据公式(1)得到的吸附能若为负值则表示SrTiO3(001)表面和N原子之间相互吸引,因此由公式(1)定义的吸附能反映了表面不同位置对N原子的吸附强度。我们在表1以及相应的讨论过程中采用了吸附能的绝对值,这也意味着N原子吸附能量值越大则相应表面位置吸附的N原子更为稳定。据我们所知:DFT-LDA方法易于高估材料的结合能(Binding energy),而结合能的精确度主要取决于交换-关联泛函(exchange-correlation functional)的选取形式即不同的交换-关联泛函都会导致其计算结果与结合能绝对值(实验值)存在一定的差异。关于SrTiO3表面吸附的理论研究 [18] 发现:交换关联泛函即使是分别选取LDA近似和GGA近似,相应结合能计算结果之间仅仅存在定性的差异,而这两种方法的数值结果具有相同的变化趋势和结论。本文工作主要考虑N原子吸附能和置换能的相对变化趋势而不是其绝对值的大小,因此本文研究的主要结论不会因为选择算法的差异而受到干扰。
表1给出了N在SrTiO3(001)表面不同吸附位置的吸附能以及N原子与吸附位置的距离。由表1可以看出,相对于孤立N原子而言,以N2分子作为参考时计算所得的吸附能明显减小,这说明表面吸附的孤立N原子更为稳定,也意味着随着表面吸附N原子的数量逐渐增多则形成N2分子脱离表面的可能性就越大。这是因为N-N之间化学键强度要高于N和O,所以SrTiO3粉体和薄膜中N-O键的热稳定性较低 [3] [4] [5] 。其次,对于TiO2原子层截止的SrTiO3(001)表面而言,N原子位于其间隙位置A上方时,其相应的吸附能量值和原子间距离最小,从而使得吸附在此表面上的N原子最为稳定。同理,SrO原子层截止SrTiO3(001)表面上的间隙位置B使得吸附在表面上的N原子最为稳定。这证实了实验检测到N掺杂SrTiO3薄膜中形成间隙原子N-O化学键 [3] 。
图2分别给出了对应N原子不同表面吸附位置的SrTiO3(001)超胞模型的几何优化结构。为了清晰简洁地显示表面层各原子相对位置的变化趋势,在图2中只给出了结构优化后每个模型近表面(仅仅包括两层原子)的原子构型。对于TiO2原子层截止的SrTiO3(001)表面,由于N原子的吸附引起了不同程度的表面结构畸变。当N原子吸附在Os(I)上方时,如图2(a)所示,相对于表面层上其它氧原子,Os(I)原子沿[001]方向向下移动进入表面层内部,导致吸附N原子不仅与Os(I)原子而且和两个邻近Tis(I)原子形成了共价相互作用。当N原子吸附在Tis(I)上方时,如图2(b)所示,尽管Tis(I)原子沿[001]方向向外移动稍微偏离出表面层,但是N和Tis(I)原子间距(1.92 Å)远大于N和Os(I)原子间距(1.33 Å) (图2(a)所示)。当N原子吸附在间隙位置A上方时,如图2(c)所示,尽管表面层的原子结构形变显著,但是由于间隙位置A紧邻的四个表面层Os(I)原子同时束缚N原子,因此使得N原子几乎完全占据表面间隙位置“融入(incorporate)”表面层,即吸附于此位置的N原子比表面层上其它位置更为稳定。
对于SrO原子层截止的SrTiO3(001)表面模型,吸附N原子的出现也同样引起了不同程度的表面结构

Table 1. The adsorption energies (eV) of N, and the atomic distances (Å) between the adsorbed N and different adsorption sites on SrTiO3(001) surface after structure optimizations
表1. 结构优化完成后N在不同吸附位置的吸附能以及N原子与吸附位置的距离
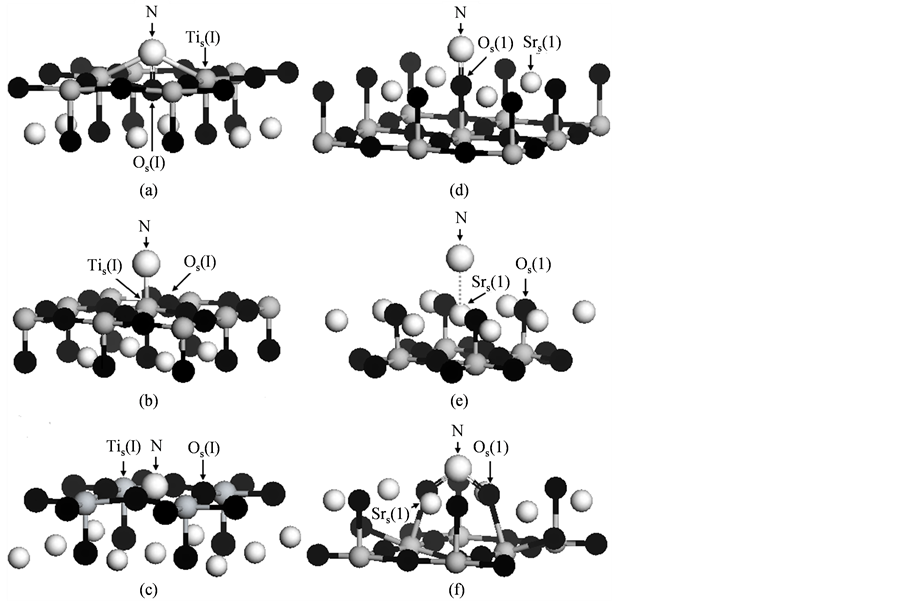
Figure 2. Optimized structures of N directly adsorbed SrTiO3(001) surface supercells, in which the unitcells are only shown in two atomic layers. With respect to the different sites, (a), (b) and (c) stand for N adsorbed on Os(I), Tis(I) and interstice A on TiO2-terminated surface layer, respectively; (d), (e) and (f) are corresponding to N adsorbed on Os(1), Srs(1) and interstice B on the SrO-terminated surface layer, respectively (where the large light gray sphere is N atom; the small light gray spheres are Sr atoms; the dark gray spheres are Ti atoms; and the black spheres are O atoms, respectively)
图2. 原子直接吸附的SrTiO3(001)表面模型优化结构示意图(在每个超胞模型中只给出了两层原子构型)。(a),(b)和(c)表示N吸附位置分别对应TiO2截止SrTiO3(001)表面层Tis(I),Os(I)和间隙位置A。(d),(e)和(f)则表示N吸附位置分别对应SrO截止SrTiO3(001)表面层的Srs(1),Os(1)和间隙位置B (其中,较大浅灰色球表示N原子,较小浅灰色球代表Sr原子,深灰色球体表示Ti原子,黑色球体代表O原子)
畸变。当N原子吸附在Os(1)上方时,如图2(d)所示,相对于其它表面层O原子,Os(1)原子沿[001]方向稍微进入表面层内部,N原子与Os(1)原子形成了共价相互作用。当N原子吸附在Srs(1)上方时,如图2(e)所示,N和Srs(1)原子间距(2.46 Å)远大于N和Os(1)的原子间距(1.35 Å) (图2(d)所示);与此同时,我们没有看到类似于N和Tis(I)原子间共价相互作用(图2(b)所示)。当N原子吸附在间隙位置B上方时,如图2(f)所示,表面层原子结构同样发现显著地晶格畸变:两个邻近的Os(1)原子明显的偏离出表面层趋向于靠近N原子,而两个邻近的Srs(1)原子偏离程度非常微弱。由于间隙位置B附近有两个Os(1)原子同时束缚N原子,因此使得吸附于此位置的N原子比其它位置更为稳定。
3.1.2. N原子置换吸附SrTiO3(001)表面
对于N置换吸附的表面模型,N原子的置换能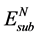 可由下式求出:
可由下式求出:
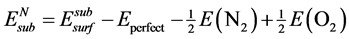 (2)
(2)
式中: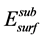 与
与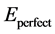 分别表示N置换吸附表面和理想表面模型结构优化的总能。E(N2) (E(O2))则表示一个孤立N2 (O2)分子的自旋极化总能 [19] 。考虑孤立原子时,则在公式(5.2)中分别用E(N) (E(O)) [21] 代替1/2E(N2) (1/2E(O2))。
分别表示N置换吸附表面和理想表面模型结构优化的总能。E(N2) (E(O2))则表示一个孤立N2 (O2)分子的自旋极化总能 [19] 。考虑孤立原子时,则在公式(5.2)中分别用E(N) (E(O)) [21] 代替1/2E(N2) (1/2E(O2))。
表2列出了N原子置换表面模型中不同位置O原子所需的能量。相对孤立的N原子而言,当以N2分子作为参考时,置换过程所需能量有所提高即形成N替位原子的可能性有所降低。对于TiO2原子层截止的SrTiO3(001)表面模型,N原子置换Os(I)原子所需能量相对较小,这意味着N原子易于置换表面层Os(I)原子。对于SrO原子层截止SrTiO3(001)表面也发生了类似现象,即表面层Os(1)原子最有可能被N原子所替代。通过分析N置换吸附表面优化结构的特征(文中暂未给出具体示意图),可以发现N原子稍稍向外偏离出表面层。这不仅与N原子掺杂TiO2表面体系的理论观点一致 [22] ,并与Ti-N化学键长度大于SrTiO3晶体材料中Ti-O键长的实验结果完全相符 [23] 。
为了比较N原子置换SrTiO3材料内部和表面氧原子的相关能量,我们建立了2 × 2 × 2的SrTiO3超胞模型并进行几何结构优化,然后仿照公式(2)求出N原子的置换能。当N原子置换SrTiO3内部O原子时,其所需能量为4.89 eV,稍大于N原子置换SrTiO3(001)表面层Os(I)或Os(1)原子所需的能量(4.49 eV或4.57 eV),二者差距非常微小(<0.4 eV)。因此,N原子是易于置换SrTiO3材料内部还是SrTiO3(001)表面层的氧原子还需要进一步考虑其它因素的影响,如热力学效应等。
3.2. 表面电子结构
为了能够更好地理解N吸附SrTiO3(001)表面的电学性质和结构特征,我们分析N原子吸附表面模型(包括直接吸附和间接吸附)结构优化后表面层上各原子的投影态密度。
3.2.1. N原子直接吸附SrTiO3(001)表面
图3给出了N原子吸附前后TiO2原子层截止的SrTiO3(001)表面层各原子的投影态密度。对于理想表面模型,如图3(a)所示,Tis(I) 3d和Os(I) 2p轨道电子的态密度分布情况与SrTiO3晶体材料的电子结构 [24] [25] 完全相符。但是如图3(b)所示,对于N吸附在Tis(I)上方的表面模型,与理想表面进行比较可知Os(I)原子的价带结构变化较小,但是Tis(I) 3d轨道电子态在价带顶的结构显著改变。恰恰是因为Tis(I) 3d和N 2p轨道电子之间的杂化相互作用,导致Tis(I)原子偏离表面层向外移动(如图2(b)所示)。对于N吸附在间隙位置A上方的表面模型,如图3(c)所示,与吸附在Tis(I)上方N原子比较,N 2p轨道电子价带宽度明显增大;此外,N 2p轨道电子态在Tis(I)导带顶上方产生了具有扩展尾翼的附加狭窄宽化峰。对于N吸附在Os(I)上方的表面模型,如图3(d)所示,Tis(I)3d和Os(I) 2p轨道电子态的尾翼扩展延伸过费米能级
Table 2. Substitution energies (eV) at different N-substitution sites after structure optimizations
表2. 表面模型结构优化后求得的N原子位于不同位置的置换能
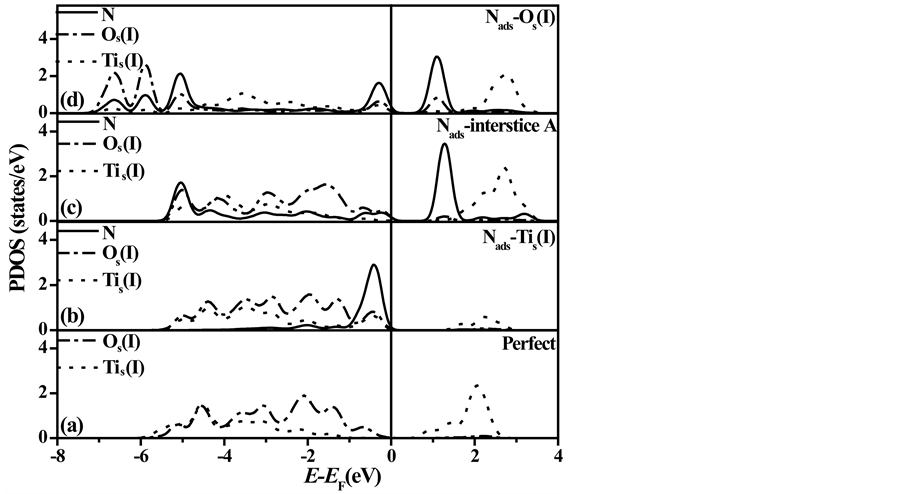
Figure 3. Projected density of states (PDOSs) of different atoms on the surface layer in N directly adsorbed TiO2-terminated SrTiO3(001) surface
图3. N直接吸附在TiO2截止的SrTiO3(001)表面层上时各原子的投影态密度
EF,而且在能量范围−7.3~−5.6 eV之间形成了一些附加狭窄宽化峰,从而导致二者的价带宽度明显增大。此外,N 2p轨道电子态在能量范围−7.3~−5.6 eV之间也形成了一些附加的狭窄宽化峰;与吸附在间隙位置A上方N原子比较,其导带和价带宽度进一步展宽。综上所述,我们发现N和Os(I)原子的杂化相互作用强度对应不同吸附位置将按照从Tis(I)→间隙位置A→Os(I),即按图3中从(b)→(c)→(d)的先后顺序逐渐增强。必须强调指出:虽然吸附在间隙位置A上方的N原子与其邻近Os(I)原子的距离(1.87Å)要大于N原子吸附在Os(I)上方时的距离(1.33Å),但是吸附于间隙位置A上方的N原子相对更为稳定(详见表1中吸附能及原子间距)。这是因为间隙位置A紧邻的四个Os(I)原子(如图2(c)所示)通过Os(I)和N 2p轨道电子之间的杂化效应同时对N原子产生束缚效应,从而使得N原子几乎占据了表面间隙位置。
图4给出了N吸附前后SrO原子层截止的表面层各原子的投影态密度。通过比较可知,SrO原子层截止SrTiO3(001)表面层上各原子的投影态密度的变化趋势与TiO2截止SrTiO3(001)(如图3所示)完全相

Figure 4. Projected density of states (PDOSs) of different atoms on the surface layer in N directly adsorbed SrO-terminated SrTiO3(001) surface
图4. N直接吸附在SrO原子层截止的SrTiO3(001)表面层上时各原子的投影态密度
似,即N和Os(1)之间的电子杂化相互作用强度对应不同的吸附位置将按照从Srs(1)→间隙位置B→Os(1),即按图4中从(b)→(c)→(d)的先后顺序逐渐增强。同样值得注意的是,N原子吸附在间隙位置B上方时的表面模型最为稳定,尽管此时N与Os(1)的距离(1.68 Å)大于N原子吸附在Os(1)上方时二者距离(1.35 Å)。这是因为N原子同间隙位置B紧邻的两个Os(1)原子通过分享2p轨道电子形成杂化相互作用,使得接近表面层间隙位置的N原子稳定性增强。
综上所述,表面层各原子投影态密度的变化趋势完全符合表面模型优化结构特征,即吸附N原子的表面层上各原子相对位置的变化趋势(如图2所示),很好地解释了表1中不同吸附位置对应N原子的相对稳定性(即吸附位置的束缚强度)。具体来讲,N原子束缚在表面层间隙位置更加稳定;而表面层O原子和吸附N原子的2p轨道电子态的相互杂化效应使得其对N的束缚强度高于Ti或Sr。
3.2.2. N原子置换吸附SrTiO3(001)表面
为了相对一致的比较分析,根据表2所列出的N原子置换不同位置氧原子对应的能量,选取N置换TiO2截止SrTiO3(001)表面层和第三层上的氧原子,即Os(I)和Ord(I)两种情形来分析N置换吸附SrTiO3(001)表面的电子结构。
图5分别给出N原子置换前后TiO2原子层截止的SrTiO3(001)结构优化模型中表面层和第三层上各原子的投影态密度。如图5(a)所示,在N置换O原子之前,相对Tird(I)和Ord(I)而言,Tis(I) 3d和Os(I) 2p轨道电子之间的杂化相互作用强度较弱。理论研究结果表明 [26] [27] ,结构优化后SrTiO3(001)表面层原子结构畸变使表面层Tis(I)和Os(I)原子的相对位置明显改变,而距离中心原子层越近的原子层面其原子结构畸变程度则显著减弱,从而导致表面层原子间相互作用强度差异显著。因此,这意味着N原子倾向于置换表面层Os(I)原子。如图5(b)所示,在N置换Os(I)或Ord(I)之后,N 2p轨道电子与Tis(I) 3d轨道电子的杂化相互作用同N与Tird(I)原子十分相似;N 2p价带电子态在费米能级EF附近形成较为明显的狭窄宽化峰并且其尾翼扩展延伸过费米能级EF。

Figure 5. PDOSs of different atoms on the surface layer and the third later in the TiO2-terminated SrTiO3(001) surface (a) before N substitution, and (b) after N substitution, respectively
图5. TiO2截止SrTiO3(001)表面层和第三层上各原子投影态密度。(a)和(b)则分别对应置换前和置换后两种情形
4. 结论
本文采用第一性原理平面波赝势计算方法对N吸附SrTiO3(001)表面模型的原子构型与电子结构进行了系统地研究,讨论分析了N原子相对表面位置的变化对其电子结构的影响,得出如下结论:
1) N原子吸附SrTiO3(001)表面的稳定性与N原子相对表面的位置密切相关。对于直接吸附的N原子,SrTiO3(001)表面层上的间隙位置相对其它表面层位置更为稳定;对于置换吸附的N原子,在不考虑其它影响因素的情况下,SrTiO3(001)表面层上O原子更容易被N原子所替代。
2) 对于N置换吸附的SrTiO3(001)表面,N原子的2p轨道电子态主要贡献于表面体系的价带电子态并且最靠近表面层O原子的2p轨道电子价带的上边缘。这一结果,不仅与N掺杂TiO2体系 [22] 的理论分析十分相似,而且和N掺杂SrTiO3薄膜的实验研究 [3] 完全相符。
目前,已有关于N掺杂TiO2材料研究发现 [22] [28] [29] :不仅N原子和O空位缺陷经常在TiO2材料内部(表面)共存,并且N原子和O空位缺陷之间的相互作用对N掺杂TiO2表面材料的电子结构影响显著,导致N掺杂TiO2表面吸附Au原子的电学性质和催化性能明显改善。同时,Shkabko等研究发现 [30] 在SrTiO3-xNy/SrTiO3-δ层状结构材料中,N原子和O空位缺陷同时存在并对其电子输运性能的影响非常显著。因此,本文工作有助于进一步研究N原子和O空位缺陷复合效应对SrTiO3材料光电性能的影响,从而指导设计N掺杂SrTiO3材料相关的电子元器件。
基金项目
本研究工作获得国家自然科学基金资助:钡锌铌系微波材料微观晶体缺陷与结构和性能相互作用的机理研究(No. 51172053)。
文章引用
房丽敏,赵可沦. N吸附SrTiO3(001)表面电子结构的第一性原理研究
Investigation of Electronic Structures of SrTiO3(001) Surface with N Adsorption by First-Principles[J]. 材料科学, 2017, 07(01): 8-18. http://dx.doi.org/10.12677/MS.2017.71002
参考文献 (References)
- 1. Wagner, F.T. and Somorjai, G.A. (1980) Photocatalytic Hydrogen-Production from Water on Pt-Free SrTiO3 in Alkali Hydroxide Solutions. Nature, 285, 559-560. https://doi.org/10.1038/285559a0
- 2. Wang, J.S., Yin, S., et al. (2004) Photo-Oxidation Properties of Nitrogen Doped SrTiO3 Made by Mechanical Activation. Applied Catalysis B: Environmental, 52, 11-21. https://doi.org/10.1016/j.apcatb.2004.03.008
- 3. Mi, Y.Y., Wang, S.J., et al. (2006) Effect of Nitrogen Doping on Optical Properties and Electronic Structures of SrTiO3 Film. Applied Physics Letters, 89, Article ID: 231922. https://doi.org/10.1063/1.2403181
- 4. Liu, C.M., Zu, X.T. and Zhou, W.L. (2007) Photoluminescence of Nitrogen Doped SrTiO3. Journal of Physics D: Applied Physics, 40, 7318-7322. https://doi.org/10.1088/0022-3727/40/23/011
- 5. Marozau, I., Shkabko, A., Dinescu, G., et al. (2009) Pulsed Laser Deposition and Characterization of Nitrogen-Subs- tituted SrTiO3 Thin Films. Applied Surface Science, 255, 5252-5255. https://doi.org/10.1016/j.apsusc.2008.07.159
- 6. Herger, R., Willmott, P.R., Bunk, O., et al. (2007) Surface Structure of SrTiO3(001). Physical Review B, 76, Article ID: 195435. https://doi.org/10.1103/PhysRevB.76.195435
- 7. Kareev, M., Prosandeev, S., Liu, J., Gan, C., Kareev, A., Freeland, J.W., Xiao, M. and Chakhalian, J. (2008) Atomic Control and Characterization of Surface Defect States of TiO2-Terminated SrTiO3 Single Crystals. Applied Physics Letters, 93, Article ID: 061909. https://doi.org/10.1063/1.2971035
- 8. Radovic, M., Lampis, N., Granozio, F.M., et al. (2009) Growth and Characterization of Stable SrO-Terminated SrTiO3 Surfaces. Applied Physics Letters, 94, Article ID: 022901. https://doi.org/10.1063/1.3052606
- 9. Enterkin, J.A., Subramanian, A.K., Russell, B.C., et al. (2010) A Homologous Series of Structures on the Surface of SrTiO3(110). Nature Materials, 9, 245-248. https://doi.org/10.1038/nmat2636
- 10. Baniecki, J.D., Ishii, M., Kurihara, K., Yamanaka, K., Yano, T., Shinozaki, K., Imada, T., Nozaki, K. and Kin, N. (2008) Photoemission and Quantum Chemical Study of SrTiO3(001) Surfaces and Their Interaction with CO2. Physical Review B, 78, Article ID: 195415. https://doi.org/10.1103/PhysRevB.78.195415
- 11. Lin, F., Wang, S., Zheng, F., et al. (2009) Hydrogen-Induced Metallicity of SrTiO3(001) Surfaces. Physical Review B, 79, Article ID: 035311. https://doi.org/10.1103/PhysRevB.79.035311
- 12. 房丽敏, 赵可沦. In掺杂SrTiO3导电薄膜光学性质的第一性原理研究[J]. 材料科学, 2015(5): 92-102.
- 13. Carrasco, J., Illas, F., Lopez, N., et al. (2006) First-Principles Calculations of the Atomic and Electronic Structure of F Centers in the Bulk and on the (001) Surface of SrTiO3. Physical Review B, 73, Article ID: 064106. https://doi.org/10.1103/PhysRevB.73.064106
- 14. Payne, M.C., Teter, M.P., Arias, T.A. and Joannopoulos, J.D. (1992) Iterative Minimization Techniques for ab Initio Total-Energy Calculations: Molecular Dynamics and Conjugate Gradients. Review Modern Physics, 64, 1045-1097. https://doi.org/10.1103/RevModPhys.64.1045
- 15. Segall, M.D., Lindan, P.J.D., Probert, M.J., et al. (2002) First-Principles Simula-tion: Ideas, Illustrations and the CASTEP Code. Journal of Physics: Condensed Matter, 14, 2717-2744. https://doi.org/10.1088/0953-8984/14/11/301
- 16. Hohenberg, P. and Kohn, W. (1964) Inhomogeneous Electron Gas. Physical Review B, 136, 864-871. https://doi.org/10.1103/PhysRev.136.B864
- 17. Kohn, W. and Sham, L.J. (1965) Self-Consistent Equations Including Exchange and Correlation Effects. Physics Review A, 140, 1133-1138. https://doi.org/10.1103/PhysRev.140.A1133
- 18. Casek, P., Finocchi, F. and Noguera, C. (2005) First-Principles Study of Oxygen-Deficient SrTiO3 and MgO(100) Interfaces. Physical Review B, 72, Article ID: 205308. https://doi.org/10.1103/PhysRevB.72.205308
- 19. Chen, Q.L., Tang, C.Q. and Zheng, G. (2009) First-Principles Study of TiO2 Anatase (101) Surfaces Doped with N. Physica B, 404, 1074-1078. https://doi.org/10.1016/j.physb.2008.11.032
- 20. Ricci, D., Bano, G., Pacchioni, G. and Illas, F. (2003) Electronic Structure of a Neutral Oxygen Vacancy in SrTiO3. Physical Review B, 68, Article ID: 224105. https://doi.org/10.1103/PhysRevB.68.224105
- 21. Baranov, A.N., Stepanyuk, V.S., Hergert, W., et al. (2002) Full-Potential KKR Calculations for MgO and Divalent Impurities in MgO. Physical Review B, 66, Article ID: 155117. https://doi.org/10.1103/physrevb.66.155117
- 22. Valentin, C.D., Finazzi, E., Pacchioni, G., et al. (2007) N-doped TiO2: Theory and Experiment. Chemical Physics, 339, 44-56. https://doi.org/10.1016/j.chemphys.2007.07.020
- 23. Schoenberg, N. (1954) An X-Ray Investigation on Ternary Phases in the Ta-Me-N Systems (Me = Ti, Cr, Mn, Fe, Co, Ni). Acta Chemica Scandinavica, 8, 213-220. https://doi.org/10.3891/acta.chem.scand.08-0213
- 24. Benthem, K.V., Elsässser, C. and Fernch, R.H. (2001) Bulk Electronic Structure of SrTiO3: Experiment and Theory. Journal of Applied Physics, 90, 6156-6164. https://doi.org/10.1063/1.1415766
- 25. Arai, M., Shigemi, K., Hideki, Y., et al. (2002) Photoelectron Energy-Loss Functions of SrTiO3, BaTiO3, and TiO2: Theory and Experiment. Physical Review B, 65, Article ID: 085101. https://doi.org/10.1103/PhysRevB.65.085101
- 26. Kim, Y.S., Kim, J., Moon, S.J., et al. (2009) Localized Electronic States Induced by Defects and Possible Origin of Ferroelectricity in Strontium Titanate Thin Films. Applied Physics Letters, 94, Article ID: 202906. https://doi.org/10.1063/1.3139767
- 27. Mackie, R.A., Singh, S., Laverock, J., Dugdale, S.B. and Keeble, D.J. (2009) Vacancy Defect Positron Lifetimes in Strontium Titanate. Physical Review B, 79, Article ID: 014102. https://doi.org/10.1103/physrevb.79.014102
- 28. Napoli, F., Chiesa, M., Livraghi, S., et al. (2009) The Nitrogen Photoactive Centre in N-Doped Titanium Dioxide Formed via Interaction of N Atoms with the Solid. Nature and Energy Level of the Species. Chemical Physics Letters, 477, 135-138. https://doi.org/10.1016/j.cplett.2009.06.050
- 29. Graciani, J., Nambu, A., Evans, J., et al. (2008) Au↔N Synergy and N-Doping of Metal Oxide-Based Photocatalysts. Journal of the American Chemical Society, 130, 12056-12603. https://doi.org/10.1021/ja802861u
- 30. Shkabko, A., Aguirre, M.H., Marozau, I., et al. (2009) Synthesis and Transport Properties of SrTiO3-xNy/SrTiO3-δ Layered Structures Produced by Microwave-Induced Plasma Nitridation. Journal of Physics D: Applied Physics, 42, Article ID: 145202. https://doi.org/10.1088/0022-3727/42/14/145202
*通讯作者。