Hans Journal of Computational Biology
Vol.4 No.02(2014), Article ID:13666,11 pages
DOI:10.12677/HJCB.2014.42003
The Visual Analysis of Coding and Non-Coding DNA Sequences
School of Software, Yunnan University, Kunming
Email: 33077511@qq.com
Copyright © 2014 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License(CC BY).
http://creativecommons.org/licenses/by/4.0/



Received: Apr. 22nd, 2014; revised: Apr. 28th, 2014; accepted: May 5th, 2014
ABSTRACT
DNA sequences include complex genetic information; their specific characteristics are contained in both the coding and non-coding sequences. Major gene components in higher levels of organisms are composed of non-coding sequences. In ENCODE project, there are evidences that 98% of the human genomes are non-coding forms and 80% of them with functions, so the research on coding region and non-coding region has become an important research hotspot. This paper provides models and experiment results which using visual representation techniques to distinguish differences between coding and non-coding sequences. This model uses probability measurements on the DNA sequences to coding and non-coding regions respectively to distinguish patterns identified from different sequences.
Keywords:Non-Coding Sequences, Graphic Representation Technique, Probability Measurements

编码和非编码DNA序列的可视化分析
刘玉倩,郑智捷
云南大学软件学院,昆明
Email: 33077511@qq.com
收稿日期:2014年4月22日;修回日期:2014年4月28日;录用日期:2014年5月5日

摘 要
DNA序列作为一种复杂的遗传信息,其具体特性不仅体现在编码序列之中,也包含在非编码序列之中。在高等生物体中主要基因成分为非编码序列,在ENCODE计划中,有证据表明,在人类基因中有98%为非编码形式,其中80%具有功能性,所以对编码区和非编码区的研究已经成为一类重要研究热点。本文提供的模型和实验结果,使用图形表示方法对编码区以及非编码区基因的差异进行区分。该模型采用的是对编码区以及非编码区的DNA序列进行分段概率测量,从而对不同的基因特征分布进行比较。
关键词
非编码序列,图形表示方法,概率测量

1. 引言
人类基因组计划(HGP)[1] 测序结果表明,DNA编码序列占人类基因组的2%,剩余的98%的均为非编码基因。非编码区广泛存在于真核生物中,众多的非编码区在生命活动中具有广泛的调控作用。ENCODE计划[2] 提供了详尽的非编码功能单位的功能图谱,在98%的非编码基因中有80%是功能性的[3] 。数据表明,在非编码功能单位中富含突变体,有时突变体出现在与特定性状相关的特异细胞中,这说明这些区域可能与疾病相关。从生物进化的观点看来,随着生物体功能的完善和复杂化非编码区序列明显增加的趋势表明:这部分序列必定具有重要的生物功能。普遍的认识是,它们与基因在四维时空的表达调控有关。因此寻找这些区域的编码特征以及信息调节与表达规律是生物信息学[4] 的重要研究内容。
面对基因组计划所得到的海量生物学数据,如何分析并从中获得生物学信息是后基因组时代[5] 的首要任务。DNA序列作为一种遗传语言,不仅体现在编码区的序列之中,而且隐含在非编码区[6] 的序列之中。基因编码区就是能够翻译成为某种蛋白质的DNA序列区域(也就是基因)。近年来完整基因组的研究表明,在细菌这样的微生物中非编码区只占整个基因组序列的10%到20%。而高等生物和人的基因组中非编码区都占到基因组序列的绝大部分[7] 。
DNA序列分析主要是分析序列中所表达的结构和功能的生物信息。其研究内容非常丰富,如:序列比较[8] 、基因识别等,而图形表示是最近发展起来的应用在DNA序列分析方面的强有力的可视化工具[9] ,它能够揭示蕴藏在DNA序列中的结构和功能的生物信息,可视化分析在人类基因组计划中扮演着重要的角色。
面对海量DNA数据,用传统方法研究这些数据,已经不能满足我们的需要。图形表示方法可以直观有效的完成DNA序列分析,相对于传统方法来说,可以缩短研究的进程。图形表示方法是对DNA序列进行分析的一种工具[10] ,在人类基因组计划中,已经成功应用图形表示方法,对DNA进行分析处理并得到DNA图谱。但是,人类基因组的可视化模式并不适用对编码以及非编码区进行分析,本文提出一种新的方法,它主要应用数学统计的原理,对DNA数据进行处理,从而得到可视化结果,对编码以及非编码区的基因分布特性进行分析。
2. 系统架构
在本节中,讨论了系统架构及其组成部分的使用图,定义了测量模型中的公式以及相关变量。
2.1. 体系结构
在本文的体系中包含有三个部分,分别是DNA概率测量映射,坐标位置映射,以及图形投影。如图1。
读取DNA序列,选择N个基因作为选定的DNA序列,作为输入数据。经过概率测量映射模块,得到归一化的概率测度。每个碱基的归一化测度对应计数均为0至1间的一个百分比,将该数据作为坐标位置映射部分的输入,按照一定的规则,对其进行处理,最终得到每个点的横纵坐标。通过横纵坐标值可以确定该点在笛卡尔坐标中的位置,将每个坐标点作为图形投影部分的输入值,集合所有的选定的DNA序列,进行图形投影,最终得到编码以及非编码区的DNA特征分布图。
2.2. 核心模块
2.2.1. 概率测量映射
如图2所示的概率映射部分由三个模块组成:柱形图,归一化柱形图和归一化测度构成。这一部分是为了得到归一化测度,以便为后面的坐标位置映射部分提供输入值。
对概率测量映射模块,详细处理流程解释如下:
中间组:
四个碱基的概率测量,表示某一个分段含有某一个碱基的数量。
相关概率测量:四个碱基得到四柱形图的相关概率测量。
输出组:
归一化测度:四归一化柱状图的相关概率测量,表示在一个分段中某一碱基在总分组中所占的比例。
数据经过预处理,形成分段模型,对于选定的DNA序列,以各分段中所含某碱基的数量为水平坐标,概率测量相同的则进行叠加,可以得到柱形图。对柱形图进行归一化处理,形成归一化的柱形图,每个碱基的归一化测度对应计数都是0到1之间的一个数(百分比),且每一个碱基在同一分组之中的计数之和为1。
2.2.2. 坐标位置映射
如图3所示的坐标位置映射部分,该模块的详细流程描述如下:

Figure 1. Architecture
图1. 体系结构

Figure 2. Probability measurement
图2. 概率测量

Figure 3. Coordinate position map
图3. 坐标位置映射图
输入组:
归一化测度:四归一化柱状图的相关概率测量,表示在一个分段中某一碱基在总分组中所占的比例。
可变参数k:一个可变的控制参数,n > 0 nÎZ。
输出组
四个成对的X-Y坐标值。
这一部分是为了得到点的坐标,从而进行投影映射,得到可视化结果。概率测量映射得到的归一化测度以及可变参数k作为输入信号,在给定的规则下完成坐标映射。最终生成的是不同参数条件下的X-Y坐标。
2.2.3. 图形投影
如图4所示的图形投影部分,该模块的详细流程描述如下:
输入组:
选择所有的DNA序列,即将所有的分组一起映射到DNA特征分布图。
四个成对的X-Y坐标值。
输出组:
四个二维的DNA特征分布图。
这一部分是为了得到最终的DNA特征分布图。在坐标位置映射部分产生的坐标值作为基本输入,但由于一个分组只能产生一对坐标值,因此收集所有选定的DNA,经过图形投影,得到DNA特征分布图。
3. 详细描述
3.1. 参数说明
n一个分段中的元素数量,n > 0;
V象征四个DNA符号中的一个,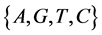 =D,
=D,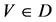 ;
;
k表示一个控制参数,k > 0;
 表示第t个分段;
表示第t个分段;
 表示具有Nj 长的第j个DNA序列
表示具有Nj 长的第j个DNA序列
 ;
;
M表示M串DNA序列;
 表示四个概率测量值
表示四个概率测量值 ;
;
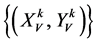 表示一组成对的坐标值,k > 0,
表示一组成对的坐标值,k > 0,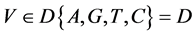 ;
;
 四个碱基的概率测量,表示某一个分段含有某一个碱基的数量;
四个碱基的概率测量,表示某一个分段含有某一个碱基的数量;

Figure 4. Graphical projection
图4. 图形投影
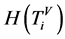 四柱状图相关概率测量,
四柱状图相关概率测量, ;
;
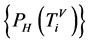 四归一化柱状图的概率测度,表示在一个分段中某一碱基在总分组中所占的比例,即
四归一化柱状图的概率测度,表示在一个分段中某一碱基在总分组中所占的比例,即

3.2. 概率测量
全部的DNA序列,分成长度为N的j个分组,将N个DNA序列分为每一段包含固定n个元素数量的t个分组。
在第一段至第t段,让0 < i < t,利用统计原理,每个分段之中某一碱基的含量为 ,
,
则有 ,每个分段序列的概率为
,每个分段序列的概率为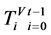 ,由此可以得到柱状图分布,记为
,由此可以得到柱状图分布,记为 。它满足条件:
。它满足条件:
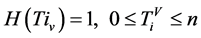
收集全部的,就可以建立柱状分布图
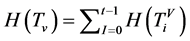
在这个条件下,四个碱基的概率为 。
。
四个向量进行归一化为
 。
。
至此,四个碱基形成了完整的测量向量。
3.3. 坐标位置映射
使用上述的测量向量,可以用两个映射函数计算的值来映射到一个二维从而可以分析DNA序列。
让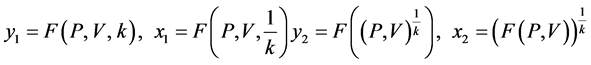
 是由以下方程定义的两组值,
是由以下方程定义的两组值,
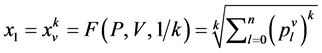
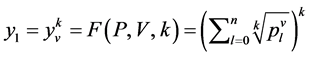


成对的坐标值分别在直角坐标系中的特定位置形成一个点,从而可以形成二维的DNA特征分布图。
3.4. 图形投影
确定选定的DNA序列(即 ),在直角坐标系中的特定位置只产生一个坐标点,所以有必要应用相对大量的DNA序列作为输入来产生可见的分布。这种类型的操作在图形投影之中完成。
),在直角坐标系中的特定位置只产生一个坐标点,所以有必要应用相对大量的DNA序列作为输入来产生可见的分布。这种类型的操作在图形投影之中完成。
对于每个分段均按照上述的规则进行操作,每个分组 得到一个坐标点,j个分组则会得到j个坐标点,将这j个坐标点映射到同一个坐标系之中,可以得到四个碱基的DNA特征分布二维图。同时,对于同一生物的编码区以及非编码区,还要进行数据量以及坐标的统一,将各个变量进行统一,以免对结果造成影响。
得到一个坐标点,j个分组则会得到j个坐标点,将这j个坐标点映射到同一个坐标系之中,可以得到四个碱基的DNA特征分布二维图。同时,对于同一生物的编码区以及非编码区,还要进行数据量以及坐标的统一,将各个变量进行统一,以免对结果造成影响。
4. 示例结果
4.1. 样品结果
利用DNA序列编码以及非编码区的文件,在两个可控的参数下(分段长度n,k)可以形成二维的图形,采用控制变量的方法,可以得到分段长度n以及可变参数k分别取值为多少时,可以得到较好的可视化效果。
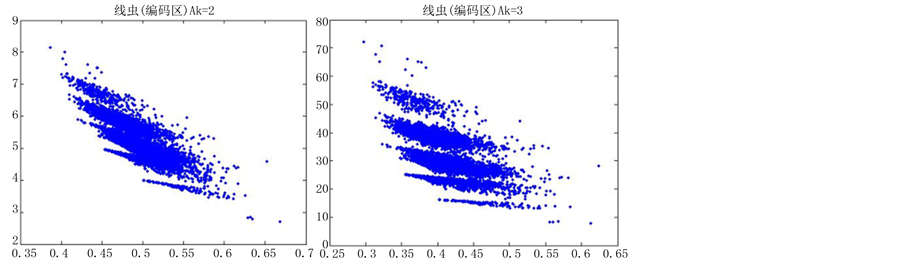
如图5(选用样品为线虫编码区碱基A)。
由上图所示,图5表示分组长度N = 500,分段长度固定为10,选用不同的可变参数k所得到的DNA特征分布图。图(a)至图(f)分别表示k值为 的可视化效果。观察各个图的聚类效果,当k = 4时的图形投影效果较好。
的可视化效果。观察各个图的聚类效果,当k = 4时的图形投影效果较好。
图6(a)~图6(f)为k值确定,分段长度不固定, 时的投影图。
时的投影图。
 (a)
(a)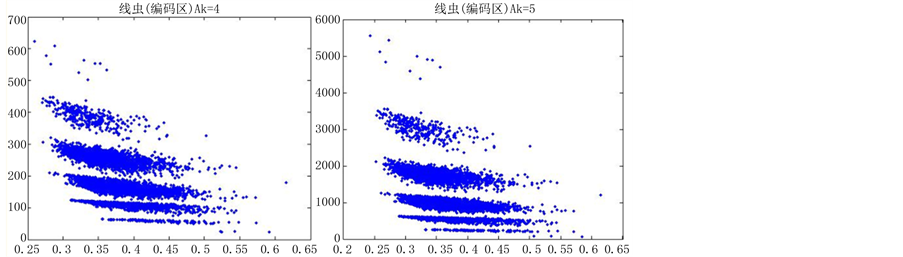 (b)
(b)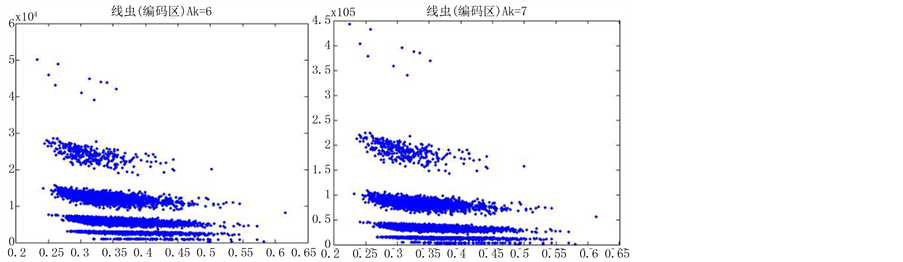 (c)
(c)
Figure 5. The variable results map of k value
图5. k值可变结果图
 (a)
(a) (b)
(b)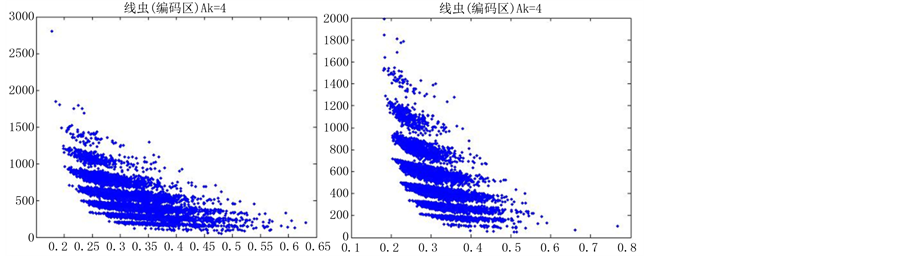 (c)
(c)
Figure 6. The variable results map of n value
图6. n值可变结果图
由上图所示,图6表示分组长度N = 500,可变参数固定为4,选用不同的分段长度n所得到的DNA特征分布图。图(a)至图(f)分别表示n值为 的可视化效果。观察各个图的聚类效果,当n = 10时的图形投影效果较好。
的可视化效果。观察各个图的聚类效果,当n = 10时的图形投影效果较好。
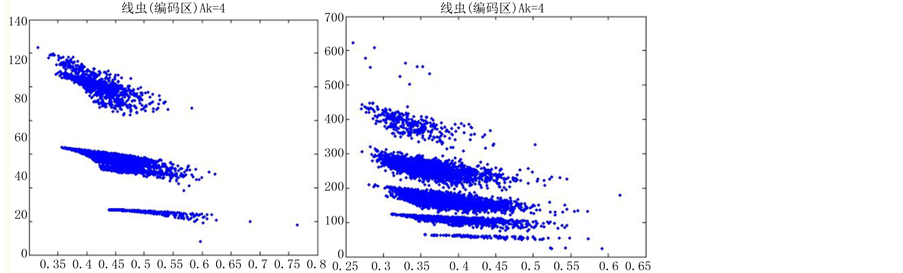
当n = 5时,改变K的值,效果图7:
在n = 5时,由于分段长度较小,投影效果随着参数的改变并没有明显的差异,不适宜用于分析编码区与非编码区可视化的分析,故文中选用分段长度(即n)为10进行可视化分析。
4.2. 结果分析
本文对拟南芥,玻璃海鞘,水稻,线虫,大肠杆菌,沙门氏菌,幽门螺杆菌以及黑猩猩的编码以及非编码进行可视化。结果图示本文以进化程度较高的线虫,黑猩猩以及比较低等的沙门氏菌为例。
 (a)
(a)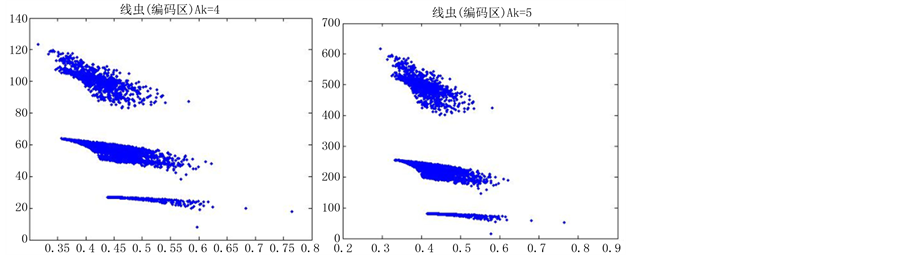 (b)
(b) (c)
(c)
Figure 7. The variable results map of k value at n = 5
图7. n = 5 k可变结果图
 条件下的可视化效果。
条件下的可视化效果。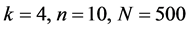 条件下的可视化效果。
条件下的可视化效果。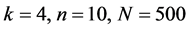 条件下的可视化效果。
条件下的可视化效果。
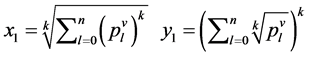
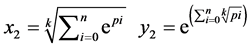
从DNA基因分布图可以看出,采用控制变量的方法,只有一个参数可变的情况下,DNA特征分布图的可视化效果有显著的差别。分段长度为10时,k值从2取到7,图形的分层效果很明显,各个坐标点明显是从集中到分散再到集中,观察得出,k值取4时,可视化的效果是最好的(如图5所示)。k值固定为4,分段长度分别为5、10、15、20、25、30,可以观察到随着分段长度的增加,可视化图形的分层
 (a)
(a) (b)
(b)
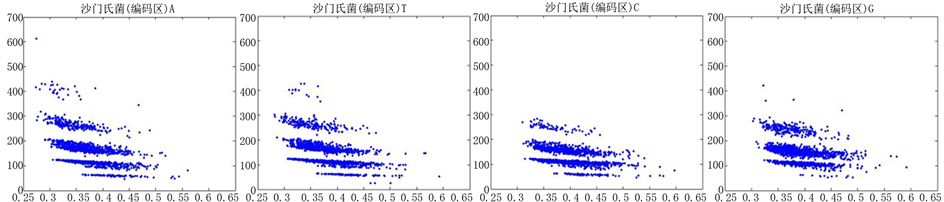
Figure 8. The visualization results of Salmonella 1
图8. 沙门氏菌编码非编码区的可视化1
 (a)
(a)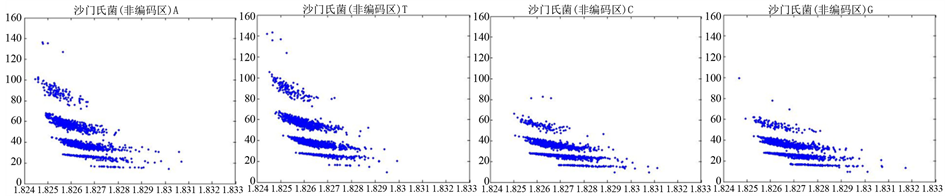 (b)
(b)
Figure 9. The visualization results of Salmonella 2
图9. 沙门氏菌编码非编码区的可视化2
 (a)
(a) (b)
(b)
Figure 10. The visualization results of Caenorhabditis elegans 1
图10. 线虫编码非编码区的可视化1
 (a)
(a) (b)
(b)
Figure 11. The visualization results of Caenorhabditis elegans 2
图11. 线虫编码非编码区的可视化2
 (a)
(a)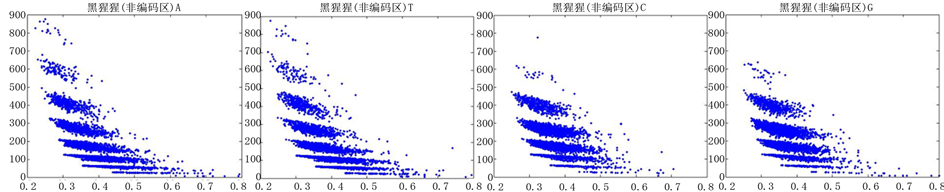 (b)
(b)
Figure 12. The visualization results of Pan_troglodytes 1
图12. 黑猩猩编码非编码区的可视化1
 (a)
(a) (b)
(b)
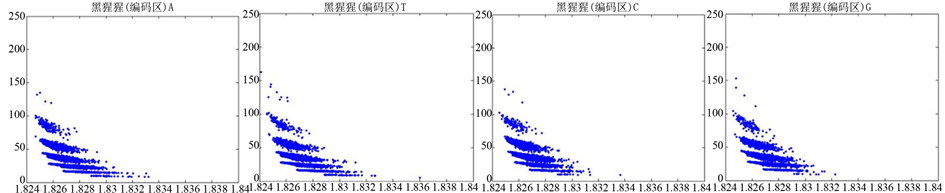
Figure 13. The visualization results of Pan_troglodytes 2
图13. 黑猩猩编码非编码区的可视化2
数量越来越多,同时也越来越紧凑,在长度为5以及10时,分层效果是最明显的(如图6所示)。
分段程度n为5时,对DNA数据进行可视化,DNA特征分布图并不随着参数的改变而有明显的改变(如图7所示),反复试验多组数据,仍得到这个结论,原因可能是因为分段长度过小,从而隐藏DNA特征分布。
观察沙门氏菌编码以及非编码的可视化效果(如图8,图9所示),两类图均存在A-T对称,C-G对称,但是总体看起来A-T比G-C在非编码投影上要明显的多。但是两类图之间,并没有明显的区别。
观察线虫编码以及非编码的可视化效果(如图10,图11所示),同样存在两类图在A-T对称,C-G对称,且A-T比G-C在非编码投影上要明显的多。与沙门氏菌不同的是,线虫的非编码区与编码区之间存在着明显的差别,它的非编码区比编码区的分布范围要大的多。
观察黑猩猩编码以及非编码的可视化效果(如图12,图13所示),黑猩猩的非编码区与编码区之间的差异与线虫相比更显明显,它的非编码区比编码区的分布范围要大的多。另外,虽然两种方法均能显示出两类图之间的区别,但是方法2(图11)的效果比方法1(图10)更明显。
从基因而言,黑猩猩是高等动物,它在一定程度上和人类的基因存在很大的相似性,线虫是较为高等的生物,它进化层次比较高,而沙门氏菌是很低等的生物,沙门氏菌比线虫和黑猩猩要低等得多。通过对编码区与非编码区进行DNA可视化分析,可以看出线虫的非编码区与编码区有明显的区别,非编码区的分布范围更广,黑猩猩与线虫相比差异更明显些,沙门氏菌则看不出什么区别。由此可以估计,越是低等的生物其基因非编码和编码越分不清,越高等的生物其基因非编码区的可视化分布图分布范围越大。
5. 结论
本文通过对编码区以及非编码区的DNA序列分组分段处理,对每个分段进行概率测量,经过归一化处理得到归一化测度,通过改变可变参数k来进行坐标映射,将所有的选定的DNA序列进行图形投影,从而得到了编码以及非编码区的DNA特征分布图。通过比较低等生物以及高等生物编码以及非编码区的基因特征分布,对二者之间的关系,从非生物学角度提供了一定的研究价值。DNA序列的图形表示方法为研究DNA序列提供了重要手段,利用图形表示方法描述基因序列具有直观性和计算简单等优点,这样相对于传统的研究方法,大大缩短研究的进程。
致 谢
感谢云南大学软件学院、云南省软件工程重点实验室信息安全基金及郑智捷博士国家自然科学基金的支持。
参考文献(References)
- [1] Kumar, S.S. (2005) Responsibilities in the post genome era: Are we prepared? Issues in Medical Ethics, 10, 150-151.
- [2] Bernstein, B.E., Birney, E., Dunham, I., et al. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57-74.
- [3] Pennisi, E. (2012) Genomics. ENCODE project writes eulogy for junk DNA. Science, 337, 1159-1161.
- [4] Ecker, J.R., Bickmore, W.A. and Barroso, I. (2012) Genomics: ENCODE
explained. Nature, 489, 52-55.
http://www.nature.com/nature/journal/v489/n7414/full/489052a.html - [5] Randić, M., Novič, M. and Plavšić, D. (2013) Milestones in graphical bioinformatics. International Journal of Quantum Chemistry, 113, 2413-2446.
- [6] Staden, R. and Mclachlan, A.D. (1981) Codon preference and its use in identifying protein coding regions in long DNA sequences. Nucleic Acids Research, 10, 141-156.
- [7] Michel, C.J. (1986) New statistics approach to discriminate between protein coding and non-coding. Journal of Theoretical Biology, 120, 223-236.
- [8] 张春霆 (1999) 用几何学方法分析DNA序列. 中国科学基金, 3.
- [9] Li, Q.P. and Zheng, Z.J. (2010) Spatial distributions for measures of random sequences using 2D conjugate maps. Proceedings of Asia-Pacific Youth Conference on Communication (APYCC) (ISTP), Kunming, 64-69.
- [10] 张巍琼, 郑智捷 (2012) 基于不同产生机制的伪随机序列和DNA序列的随机性测量. 成都信息工程学院学报, 6, 548-555.