Journal of Advances in Physical Chemistry
Vol.07 No.01(2018), Article ID:23644,10
pages
10.12677/JAPC.2018.71002
A Quantum Chemistry Study of the Chemical Bond (1)
Guangyao Zhou
Beijing Kein Research Center for Natural Sciences, Beijing

Received: Jan. 10th, 2018; accepted: Jan. 23rd, 2018; published: Jan. 31st, 2018

ABSTRACT
Some quantum chemical concepts involved in study of chemical bonds are described. By means of quantum chemistry calculation, we conducted molecular orbital analysis and electron density difference analysis, and from view point of Hellman-Feynman electrostatic theorem, we investigated the nature of covalent bond, which is one typical kind of chemical bond. We showed that covalent bond is binding force caused by electrons in bonding region shared among nuclei (or groups).
Keywords:Chemical Bond, Covalent Bond, Hellman-Feynman Electrostatic Theorem, Bonding Region, Electron Sharing, Group
化学键的量子化学研究(一)
周光耀
北京科音自然科学研究中心,北京
收稿日期:2018年1月10日;录用日期:2018年1月23日;发布日期:2018年1月31日

摘 要
阐述了化学键研究所涉及的几个基本量子化学概念。通过量子化学计算,进行分子轨道和电子密度差分析,从Hellman-Feynman静电定理的视角,考察了化学键之一共价键的本质,共价键是在核(或基团)之间共享成键区电子所引起的结合力。
关键词 :化学键,共价键,Hellman-Feynman静电定理,成键区,电子共享,基团

Copyright © 2018 by author and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 引言——基本概念
1.1. 量子化学计算在做什么?
量子化学计算,是在一个给定的原子核空间排布所形成的核势场中,安排电子使体系能量最低。
DFT (Density Functional Theory)开宗明义就说是电子密度的泛函,在算体系能量最低时的电子密度分布,认为通过体系的电子密度可得到体系一切性质(包含电子波函数y)。
MO (分子轨道)理论自洽计算化学体系的电子波函数y并求得能量,而可通过y2 = r,得到电子密度r,产生电子在原子核空间的密度分布,并选定能量最低时对应的波函数。也是在安排电子。
这样,量子化学计算就给出了化学体系中的电子结构、电子云r。然而,这电子云r是客观存在的吗?
这个问题涉及到量子力学的基本假定。整个量子力学是建筑在几个基本假定的基础上的。这些假定如同几何学中的公理一样,是不能被直接推导或证明的。但它们是从实践中归纳抽象出来的,它们的正确与否只能不断地被实践检验、证实。y和r实际上就是量子力学的第一个基本假设。因此,我们只能认为量子化学得到的电子云r是不同程度地符合客观化学实在的。y不只是为了使体系能量最低而选取的纯抽象数学函数,而是在严格、精细地安排电子,而且用电子密度r表达能量最低时电子在空间的分布,来接近真实地反映、描述电子在核势场空间的客观存在、分布。
对于一个分子轨道MO,就相应有对应的y和r。y是电子在它所处的核势场空间中作为波时的状态函数,r是电子作为粒子时在核势场空间中存在的密度分布,这是电子的波粒二相性。
任何原子、分子的化学存在和性质,都必定会有一个对应的体系的波函数y和电子密度r,任何化学变化,必然引起体系的波函数y和电子密度r的变化。没有脱离y和r的化学存在、性质和化学变化。
DFT的原理实际上与MO理论基础是相同的,只是MO理论从头计算y并得到r,而DFT在从MO理论得到一个初始的r之后,构造一个数学泛函来专门计算体系具有最低能量时的r,这样大大地减少了计算量,对大分子适用。后来又想出了另外的补充数学函数,来弥补对长程相互作用计算的不足,可以计算范德华作用体系,即DFT-D。
1.2. 电荷的困惑和出路
研究化学问题,总是想得到分子的电荷分布。其实,电荷有多种概念、层次。
第一个层次是物理有机的形式电荷,它概括了分子中由于原子参与成键的轨道电负性不同,电子会发生转移这种事实,把电荷归结到具体的原子之上,认为电负性大的原子会完全吸引过来电子形成负整数价,用−1、−2来标示,这种形式电荷在一定程度上可以方便地解释化学性质。然而这毕竟过于简化、绝对化,不能精确、真实地反映客观化学的具体存在,有时还会产生误导,不利深入定量研究。
第二个层次是有了量子化学计算之后,分子中的电荷分布可以由各种精度的方法计算出来。分子中的电荷分为原子上布居和原子之间重叠布居。这是由于MO是原子轨道的线性组合(LCAO),一平方求r,就产生了一系列原子之间的重叠布居。这时,为了方便应用,还是按照认识的惯性,将电荷划归给原子,最简单的方法是由原子双方均分重叠布居,最有代表性的布居分析是Mulliken atomic charges,即常用的净电荷。但长期的应用实践中发现它经常出现不符合实验预期的结果,后来出现了一些改进方法,较为人们接受的是NPA (自然布居分析)。然而电荷问题并没有真正解决。
Mulliken atomic charges的根本缺点在于:
1) 当LCAO中的基函数变化时,均分重叠布居引起的误差也随之变化,电荷出现不可预知的变化。
2) 人为地均分了重叠密度,对象体积相差很大的原子,误差很大。
3) 更为严重的是,分子有σ、π、n (孤对电子)、δ等各层轨道,参与这些轨道的原子轨道的电负性在分子中是发生了变化的,从而各层分别有自己的电子流动,流动方向也不相同。Mulliken atomic charges将各种轨道的电荷笼统地归结在一个原子上,自然会不准确、出问题。
由于Mulliken atomic charges是量子化学计算的经典输出,而其数值又常不合理,经常与实验结果预期不符,以至于电荷的应用被淡化、边缘化了,不再使用电荷了。精确的量子化学计算,一到电荷就出现问题了,产生所谓电荷困惑。
第三个层次是电子云的另一种计算和表达,它是分子在全空间的电子密度r的分布,不用刻意把电子归结于哪个原子,这更符合化学分子的客观实际。它是对于空间点阵的任一格点,先把LCAO在这点的值线性加和计算出来,再平方即求得r的值,从而免去了大量难解难分的重叠密度。这里,电子密度ρ就是在一定布局的原子核外的一个空间电子分布,一个几率密度分布。它可以是整个分子的,也可以是某一个或某几个MO的。而且还可以将电子密度ρ乘以它所在的体积微元得到几率,将分子存在的整个空间或部分空间的几率加和起来,就得到这种空间的电荷数值。当作电子密度差Δρ时,还可以分别对正值区和负值区进行加和,甚至对某一个选定的局部空间(如某个指定原子空间)的电荷增减值分别加和,从而得到分子中电荷的净变化的确切数值。
应该恢复电子密度r作为量子化学第一手原生态信息地位,认为量子化学计算就是在核之间精细地安排电子、共享电子。电子、电荷通常是同义语,但有时对空间具体的电子计算数值称为电荷。
一个MO的ρ,是电子在空间分布的写照,没有人为的划分和失误。据此可以得到电子分布的信息。然而ρ是那么浑圆、囫囵,它是一片云。
在量化图形中,电子密度差(electron density difference) Δρ值得重视。它其实是传统的原子净电荷概念的扩大和空间化,它直接从MO、ρ衍生而来,真实地、科学抽象地表现了电荷在分子空间分布的净变化,并将它展示出来,不再只将净电荷人为地局限、归并在各个原子上。其实在分子,更应该关注的正是原子核之间的电子分布状况和量值,不应该将重叠布居人为地划分给原子、均分了事。
一旦作出了电子密度差Δρ图形,成键原子前后变化、分子之间氢键、络合、范德华作用、吸附等,电子增减的净变化,被鲜明地、细微地突出了,可以发现许多新东西、意想不到的东西。
成千上万的精确的电子密度ρ值以及据此作的Δρ图,一旦作成了图像,似乎又失去了数值的精确性,在更高的层次上变成定性的图形了,但这定性图形却展示了隐藏在这成千上万大数据之上新的质,并通过图形表达出来,这有助于我们认识、揭示量化计算的成千上万大数据所反映的化学本质。而面对图形,我们好像看见化学实体了,人们对图像的识别远比成千上万的数字敏感。特别是我们常常需要考察分子成键的单个特定的MO(或者孤对电子MO等),电子密度差Δρ也能表现它在分子中的电子净变化,不致将各种轨道的电子叠加在一起混为一谈。
如实地考察化学体系空间电子分布的净变化Δρ,无疑是电荷运用的出路。
1.3. 电负性和电负性均衡原理
Pauling首先提出原子电负性概念 [1] ,说电负性是成键原子的键轨道吸引电子的能力。并用同核双原子所构成的分子的实验结合能以经验公式计算原子这个成键轨道的电负性。Pauling电负性已经成为经典,至今得到广泛应用。后来原子电负性概念又自然扩大到基团电负性。
基团的概念在化学中已经习以为常,如:甲基CH3、羟基OH。从任一个化学键看,可以认为构成化学键的两边的分子片分别是两个基团,这两个基团参与成键的价轨道共享电子而成键。
在DFT,Parr等人 [2] 指出:电负性是体系电子化学势的负值。
同样将Pauling的电负性推广到MO理论,电负性χ是原子AO能级或分子MO能级ε的负值。
能级ε是从轨道上拿走一个电子所要付出的能量,电负性χ则是该轨道吸引电子的能力。这是一个客观事实的两个相反的观察视角,用一个负号表达视角的相反。电负性视角接近化学实践习惯,是人们感知的一种客观存在。
这里电负性的范围不仅只是成键的价轨道,而是所有的分子轨道,它们都有轨道的能级值,自然也有相应的轨道电负性。就是断开成基团,断开出来的轨道也是有电负性的,称为基团电负性。
不同电负性的原子组成分子时,参加MO的原子轨道(以基函数表示),通过得失部分电子调整着电子对核的屏蔽,从而调整着自己的电负性,也即调整着自己的能级。
原子之间成键时,电负性大的原子参加成键的轨道得到部分电子,电子对核的屏蔽增强,使其电负性降低,电负性小的原子失去部分电子,电子对核屏蔽减弱,其电负性升高,从而达到双方电负性均衡、不再有电子流动。两个基团结合成分子时,通过电子在基团之间转移,这两个基团电负性也是相等的。
这是电负性均衡原理企图概括的事实,它是能量最低原理的一种表现形式。水往低处流,直至均衡。
我曾经提出 [3] 在MO理论中电负性均衡原理的基本表述:在分子的一个MO中,组成这个MO的各个原子的电负性通过电子调整之后是相等的、均衡的,它的值就是这MO的电负性(能级的负值)。并作了阐述。
1.4. Hellman-Feynman静电定理的成键区
所谓核(或基团)之间的电子共享,这共享的电子实际上就是指核(或基团)之间处于“成键区”的电子,成键区的共享电子吸引着与它相关的核(或基团),对抗了核与核的排斥、电子与电子的排斥达到平衡产生了化学键。关于成键区的概念和实际区划,见Levine的《量子化学》 [4] 的论述和图示,它是根据Hellman-Feynman静电定理实际计算得出的,成键区图示本文转引见图1(A)。当我们一提到电子共享时,就要立即联想到这个成键区,是核(或基团)静电吸引着成键区的电子云。特别是当拿Δρ说事时,也不要仅限于Δρ本身,而是要想到成键电子所构成的整个空间的电子密度ρ,尽管Δρ是成键的精髓、解释成键本质的钥匙。当然,还应注意到反键区的电子区划。
Levine在论述这个静电定理时还说:作用于核的力是静电力这个事实,确证在分子中没有“神秘的量子力学”起作用。也就是说化学键也只是电子共享,是核对电子的静电作用,没有“神秘的量子力学”起作用。而现代量子化学程序对成键区的共享电子、反键区的电子作了精确的计算,由此得到了电子云分布、电子所处的能级和体系的能量。尽管原子结合成分子时是经由双方波的相互作用融汇贯通的结果,具有波的层面,但是当从核吸引电子的视角去观察时,只能得到电子云这样的粒子结果,只能是核对电子云的静电作用。当然在量子化学,核静电吸引的是电子云,电子之间的排斥也是电子云之间的排斥。
可见,化学键的物理定义应是:原子核(或基团)对成键区的电子共享产生了结合力。这结合力对抗了体系中电子之间、核之间的斥力,达到一定程度的稳定平衡。

Figure 1. (A) Binding and antibinding regions; (B) ρ in H2; (C) Δρ in H2; (D) Δρ in HF
图1. (A) 成键区和反键区;(B) H2的ρ;(C) H2的Δρ;(D) HF的Δρ
在对化学键认识的早期,在共价键的概念下,在成键原子之间画一条线表示化学键的单键,还用两条线、三条线分别代表双键、三键。在金属有机物中还有四键、五键 [5] 。后来对化学键的认识日益深入了,建立在量子化学基础之上了,这种用线表达化学键的概念实际上也随之与时俱进,不断修正、补充着新的内容,使这种表达仍不失为一种简单好用的合理科学抽象。这线条,现在实际上代表着在基团之间成键区的电子共享,只是这电子共享有着各自的形成来源和客观的存在现实形式而已。实际上远不是一条线,远不是一条线所能完全反映的,深入、精细的研究,还是要看电子云的成键区和反键区。
当我们看到这种线条就联想到那存在于成键区的种种共享电子云时,甚至看到在成键区的Δρ时,这种线条就是一种合理的科学抽象。但是,当我们在新发现的化学体系中不能用这种线条来连接原子成键,或是电子并不直接分布在二核(基团)连线之间,就认为没有化学键时,这种线条就是形而上学了。这就需要把化学键还原到基团之间的成键区电子云共享了。
1.5. 本文的一些约定
本工作用Gaussian16程序 [6] 进行量子化学计算,各具体计算方法(选择收敛好、合用的)在文中说明,而基函数统一用6-311++G**,另外的基函数在文中说明。进一步用Multiwfn 3.1 [7] 进行数据处理和作图。
作体系与各原子的电子密度差Δρ时,原子波函数的数据取自Multiwfn程序自带的原子波函数文件。等值线起始值为±0.0004,用几何级数来生成等值线,步长为2,设定等值线条数为30。虚线为ρ减少的区域,实线为ρ增加的区域。此外的数值则在文中说明。
距离不标出单位时是Å,电荷和能量数值不标出单位时是a.u. 伸缩振动频率单位为cm−1。
2. 共价键的电子共享
通常认为化学键包含共价键、离子键、金属键、多中心键。此外还有化学作用(氢键、范德华作用等)。本文从电子共享的视角深入地研讨一下共价键。
2.1. 众所周知的H2再观察
H2分子的共价键,是电子共享的典型,在各种化学键都可以找到这种电子共享的特质和形象。
用CCSD/aug-cc-pVTZ计算H2分子。计算的电子密度的截面图见图1(B)。它的计算键长为0.7426 (0.7421 [8] [9] ,括号中为公认的实验值,下同),结合能(未考虑振动零点能)为0.1730 (0.1744 [8] ),伸缩振动频率为4406.39 (4401.21 [8] [9] )。
虽然不能实验测定H2分子的电子云ρ,但是由计算出的ρ算出的上列数据与实验测定值如此相符,说明计算得到的ρ是反映了客观存在的。
上面的结合能是H2分子的能量和两个自由H原子的能量之差得到的。同样可以作H2分子的电子密度和两个自由H原子的电子密度,然后作电子密度差Δρ,见图1(C)。这是大家最为熟悉的,电子往键中间增加聚集形成成了共享电子的增加,Δρ表明电子在二核周围和中间的聚集净增加了0.2478。
从图1(C)可以看到,H原子形成H2分子时,电子中移至成键原子的之间,这是人所共知的了。现在我们的视角转向分子中的原子,由于电子中移,造成了H原子的背面有较大的电子缺失、余下的电子还向H原子核紧缩。这时分子中H因电子缺失电负性大大提高,虽然没有电子在原子之间转移,H原子也并没有保有它作为孤立原子时的电负性。这种分子中成键原子的电子大大缺失向成键原子中间聚集,是一个普遍现象,是共价键的主要特征,是原子(或者基团)成键后其能级大大下降的物理根源。
这种电子往键中间聚集形成对成键二核之间的屏蔽增大,减小了原子核与核之间的排斥力,使体系的总体能量降低。
我们说,MO图和ρ图,是用来描写MO和ρ客观存在的具体形象的,它可以说是MO和ρ存在的某种近乎真实的形象写照。粗略地说它俩就是这个样子!现在核(或基团)吸引的是电子云,电子之间的排斥也是电子云之间的排斥。
但是Δρ呢,它是客观存在吗?
我们打个比方:兄比弟大3岁,这个3岁之差,就像是Δρ,它是一个差值,它当然是客观真实存在的。兄、弟都是真实存在着实体,但是并没有什么“3岁”这个实体、对应物存在,也即并没有Δρ的实体、对应物真实地在那里单独存在,Δρ只是一种两种客观存在实体的差值、科学抽象,图形也只是这科学抽象的形象展示而已。
明白了这一点,我们才可以科学地运用Δρ所反映的真实。如果把Δρ就当成真实实体形象,并引伸出某种结论,就有可能陷于可视化图形导致的错误。
我们通常认为,对于共价键,电子会往键中间聚集,本文还说是“中移”,于是就会拿Δρ图作为有力的证明,电子果然在键中间有聚集成团的形象,于是教材上指着这个Δρ图说:共价键就是这个样子!然而这样说是片面的。为什么?
如果单看H2电子密度ρ图,见图1(B),电子聚集在核周围、且层层向外电子密度减小,直至二者电子密度相遇而弥合、贯通,交接处密度还是最低的,并没有真在键中间聚结堆积成团。这键中间的电子增加成团的图像,只是两个近乎球形的波相遇、叠加、贯通,填补了波形之间的不吻合、稀缺之处而已。
电子密度差Δρ鲜明地突出了电子成键前后电子在空间的净变化,细究起来实际上包括三个部分。
第一部分是电子中聚,电子填补稀缺、融会贯通、平滑过渡,而不是产生球形电子在键中间的堆积。
第二部分是电子向键轴方向浓集、堆积,在核(或基团)之间的连线上密度最高。中国科学院国家纳米科学中心的科研人员,在国际上首次“拍到”的氢键“照片”,它实现了氢键的实空间成像 [10] 。这是8-羟基喹啉分子之间的氢键。前文 [3] 之(一)引用过相关的照片。当时是看的氢键。现在专看8-羟基喹啉分子本身,可以看到该分子骨架共价键,它们都呈现亮线,表明电子在键轴的浓集,它是在原子之间画一条线代表价键的客观存在的物理基础,这一条线是多么好的科学抽象。
第三部分是因为电子中移、向键轴浓集,使得H有所裸露,电负性增强,剩余电子收缩浓集。
现在的Δρ把这三种电子重叠在一起了。如果用最小的基函数STO-3G来算Δρ,将只看到二H之间填补稀缺的电子聚集,H却出现在密度减少区,这样的基函数反映不了H有所裸露引起的剩余电子浓集。
与H2这个典型的例子一样,所有的共价键都是一个弥合波形之间不吻合、填补波形之间存在的稀缺,向键轴方向浓集,形成平滑、沟通、均衡的MO的过程。键中间的电子密度是增加了,但那都是平滑过渡而没有额外成团堆积的,而且还是越往核走电子密度越大。形成这种存在的原因,是电子落入核(或基团)之间的势场时所取的波函数状态或者说电子密度分布就本来是这样的。
从Hellman-Feynman静电定理来看,成键只在于共享成键区的电子,不在于电子配对所产生的所谓量子效应,电子配对只是说一个MO可以容许两个自旋相反的电子存在而已。而且这对电子在这个MO上还要因电子彼此排斥而处处远离,尤其是不能同处在一个微空间,所谓“电子相关”,就是把HF方法同处在一个微空间的误算用数学的方法加以剔除。
可见,现在讲“共价键”,只是对成键区电子共享的概念,它可以是配对电子,也可以是单电子,甚至是分数电子。共价键并不是真的在共享全部配对电子。其实,在反键区也是有许多电子分布的,成键区安排不了那么多电子,那样电子之间的排斥会太大。
2.2. 极性共价键
以极性分子HF为例,采用CCSD/aug-cc-pVTZ计算,并作零点能校正。计算得到的键长为0.91808 (实验值0.9168 [8] [9] ),结合能为:0.209267 (0.2120 [8] [9] ),伸缩振动频率为4172.10 (4138.32 [8] [9] )。
虽然不能实验测定HF分子的电子云ρ,但是由计算出的ρ算出的上列数据与实验测定值如此相符,说明计算得到的ρ是反映了客观存在的。
上面为准确计算能量用的post-HF方法,然而它有大量的小分数占据轨道,不便使用。下面看各MO和分析ρ时,是用的HF/6-311++G**的输出结果,从电荷的视角来分析问题这已经足够了。
极性分子HF,它除了中移共享电子外,还伴随着有电子在原子之间的转移。HF分子可以看作是H的单电子和F的单电子形成的共价键,并形成了HF分子的一个MO3 (即σ键)。图1(D)是HF分子的MO3与H的单电子和F的那个单电子作的电子密度差Δρ图。这是只对HF分子的MO3作Δρ,如果作全电子的Δρ,F上的孤对电子虽然与分子的成键轨道MO3是正交的、不参与成键,但是HF的键电子与孤对电子的排斥以及F上转移电子增多会使孤对电子发生形变,杂在Δρ图中影响对键电子图像的识别和分析。
从MO系数和Δρ图看,这个σ键可以分析为三个内含:原子以基函数形式参与成键、电子在不同电负性原子之间转移、电子向键中间移动(为了与原子间电子“转移”区别本文称之为“中移”)。
第一、原子以基函数组合形式参与成键,这基函数组合或者可以看作是广义的杂化。
对于成键来说,基函数是采用各种轨道线性组合,来均衡不同原子能级或电负性的差距、弥合AO之间的波形的不吻合,应对键间电子向键轴的聚集、收缩,以便融合成平滑、贯通、均衡、统一的MO。
对非键电子来说,如孤对电子n,基函数为了适应下层σ键电子增多、n能级被抬升,以及可能的反馈,基函数描述了它的膨胀极化、弥散变形。
对于吸引转移过来的电子,基函数还会“妥善安排”。
基函数是借用AO来修补成MO的,它不仅是计算需要的一组数学表达式,当它们分配了系数、也即安排了电子,它就形成了一个新的广义杂化的空间AO或MO,是具有能级值或说是电负性的,它是电子的载体、存在的空间,是电子落入该核势场中所取波的状态或电子云。
第二、电子在不同电负性原子之间转移。
从HF分子Δρ图可以清楚地看到,H缺失了大量的电子,导致它电负性升高;F增加了大量的电子,导致电负性降低,直至二者电负性相等、均衡。电荷转移量经核算为0.3644。
有趣的是,通常说sp杂化,想当然地认为会用增大的一瓣去成键以增大重叠能力。但在HF中,MO的基函数中F的s、p却是这样的杂化,即F以缩小的一瓣去成键,而用增大的一瓣去装转移过来的过多电子,这转移电子妥善安排在离键远的外面、反键区集结,以减少电子之间的排斥,它本身就是电子排斥的结果。不能想象成键时所谓共享的配对电子都出现在核(或基团)中间的成键区,那样电子之间会安排过密、排斥过大。
对照图1(A)和图1(D)看转移电子,它正好符合反键区所划出的形象范畴,说明转移电子是反键电子,并不会形成所谓极性键因正负电荷分离、产生静电吸引对成键作贡献。
第三、电子向键中间中移。
从HF分子Δρ图看,在H和F之间也有明显的电子中移,经核算Δρ中移的电荷为0.1890。这与H2分子的情形完全一样,它同样会造成H和F的电子因中移而有所缺失,这从中移的电子增值两边同样有减少可以看出来。只要成键,这种电子中移而使成键双方缺失电子是普遍存在的。只是这种电子中移与电子转移叠加在一起,H更加缺失电子了,而F的因中移造成的电子缺失被过多的转移电子增加掩盖了。但F的前面也有虚线在表征电子的缺失。当然F的中虚,一方面是由于电子中移,另一方面是由于大量的电子转移过来,电子之间排斥增大,造成电负性降低,电子层膨胀引起的。这里MO3 (即σ键)的Δρ,省去了孤对电子的变化。
电子往键中间聚集、形成电子对二核的屏蔽,会减小核之间的排斥力,有利于降低体系的总体能量。
这里,通过转移电子、均衡不同原子能级或电负性的差距,应对键间电子向键轴的聚集、收缩,弥合AO不吻合的波形,从而使AO融合成平滑、均衡、统一的MO,都是通过精心选择、分配基函数系数来完成的,即是在精细地安排电子使体系能量最低。
如果从电负性这个角度看过去,量子化学在作什么呢?量子化学在原子组成的分子时,通过调整原子上的电子量使原子的价轨道电负性均衡(当然这是通过波函数基函数的精心分配系数进行的),从而形成统一融合的MO,并且安排了电子,就这样一层层地安排电子。
所谓电子在原子之间转移,无非是说电子云多倾向某原子一些而已,所谓电子中移,无非是说弥合的电子云均衡地填补了中间的稀缺之处而已。
只有在成键区精心安排处于能量最低的电子,才是此处极性键的共享的电子,不仅是Δρ展示出的。
2.3. 配位键
凡共用电子对是由一个原子单方面提供而形成的共价键称为配位键。配位键典型的例子如H3N→BF3 [11] 。
H3N→BF3用CCSD/6-311++G**进行优化计算,优化后N-B的距离为1.66698 (实验值1.60,是晶体中的实测平均数据 [11] ),N、B共价半径之和为0.71 + 0.65 = 1.56。配位键要比相应的通常共价键要弱 [11] ,故比共价半径之和微长。零点能校正后的两个基团H3N、BF3中N→B结合能为0.069493,N-B的伸缩振动频率为692.56。计算值与实验值基本符合,说明计算得到的ρ是反映了客观存在的。
算结合能时对H3N和BF3也取配合物中的构型,不进行优化。因为BF3的优化会成为平面,在平面时B的p型空轨道与F孤对电子p型轨道相位相符形成大π键 [12] [13] ,能量下降很低,与在H3N→BF3中的电子结构不相对应。
下面用输出的HF/6-311++G**结果分析它的MO和ρ。
在H3N→BF3的MO中,MO21(HOMO)即是N→B配位键的形象,它的截面图见图2(A),虚实线是相位。这个截面上刚好不包含3个F和3个H,只看N→B。图中除了表达N的孤对电子向B的空轨道配位外,还表达了N的孤对电子与三F的反相位孤对电子的排斥(三F不在这个截面,截到的是孤对电子波函数在这个截面的值)。图2(B)是MO21的电子密度r的截面图,对照图1(A),可以划分出这个配位键的共享电子的成键区。可见所谓配位电子,也并不是这两个电子都共享了,共享的只是成键区的电子。
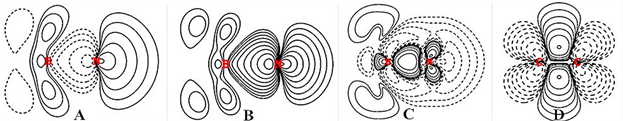
Figure 2. (A) MO21 in F3BNH3; (B) ρ of MO21 in F3BNH3; (C) Δρ in F3BNH3; (D) Δρ in CH2CH2
图2. (A) F3BNH3的MO21;(B) F3BNH3的MO21之ρ;(C) F3BNH3的Δρ;(D) CH2CH2的Δρ
还可以作H3N→BF3与组成它的H3N和BF3的电子密度差Dr,见图2(C)。这个截面上刚好不包含3个F和3个H,只看N→B。
图2(C)在正中间展示了H3N的孤对电子向BF3的偏移配位的轨迹增量,产生了一个共价键。在H3N中,三H向N提供了电子,孤对电子能级被抬高了,使它可以给出电子。在BF3,三个高电负性的F从B拉走了过多电子,使得BF3有一个很低的空轨道接受电子(即LUMO,它的形象正是B上的那个p型空轨道,但是它的能级却是有一个电子占据时计算出的,故能级很高),使H3N的孤对电子可以偏移配位过来,在成键区共享电子形成共价键。虽然这种Dr将H3N和BF3的NH、BF的成键电子密度状况基本抵消了,不会显示出来,但H3N的孤对电子偏移配位还是会引起体系电子分布的变动。如孤对电子偏移配位使B的电子增多、电负性下降,有电子进一步流向F,在图2(C)中B的左上、左下截到了F在这个截面的电子增量。而N因孤对电子偏移走了,电负性增大,N从H那里拉来更多的电子,也有显示。
从整个图看,电子向配位键轴收缩了,周围相应有电子减少。落于成键区的电子也并不是N的那个配位电子的全部,具体落入成键区的电子量还要将N的孤对电子的分布与成键区的区划对照来看。这在量子化学程序计算时已经是这样作的了。
2.4. π键的电子共享
π键也是一种电子对共享,属于共价键。最单纯的例子是CH2=CH2 [14] ,是σ键和π键构成的双键。
对CH2=CH2用CCSD/6-311++G**进行优化计算。得到C=C键长为1.339 (实验值1.33 [14] ),键角∠CHC为121.60˚ (121.3˚),∠HCH为116.80˚ (117.4˚)。结合能0.2667 (0.2724)。C=C伸缩振动频率为1689.4 (1640)。计算值与实验值基本符合,表明计算得到的ρ是符合客观存在的。
用RHF方法计算结果分析π键电子密度差Δρ。设定CH2=CH2所在的平面为xy,由于轨道对称性各别,有一个独立的MO来表达π键,可以选这个MO的波函数wfn文件。另外,对于两个分子片CH2,可取在CH2=CH2中的构型坐标分别进行计算,令它的多重度为3,它有两个单电子轨道,其中一个Pz就是将要形成π轨道的,分别作这样两个CH2的Pz的波函数wfn文件。然后作CH2=CH2的π-MO与两个CH2的Pz的电子密度差Δρ,选择xz平面(y = 0)作为截面,就得到成键前后π键电子密度差Δρ,见图2(D)。这里不包含σ键的内容。
与上面H2的σ键相类,π键电子的上下两瓣也中移至成键原子的之间,弥补了原来两个P型两瓣椭球并排重叠时的稀缺以及浓集,而相应的部位因电子中移有明显地减少,电负性提高(能级下降)。电子增加的区域正在成键区,产生一种π键的电子共享(不仅是Δρ的增量区,还包含在成键区中的ρ)。
3. 结论
化学键中的共价键是原子(基团)之间对成键区的电子共享,共享电子同时吸引着各个原子(基团),克服了各原子(基团)之间的核与核的排斥和电子与电子的排斥,达到吸引和排斥的稳定平衡,形成了化学键。
极性共价键的结合力,不简单是形成正、负离子引起的纯粹静电引力,而是共享成键区的电子。极性键的转移电子多存在于反键区,并不对成键作贡献。
致谢
本文由北京科音研究中心的卢天博士提供计算资源,并对此研究工作进行了讨论,特此致谢!
文章引用
周光耀. 化学键的量子化学研究(一)
A Quantum Chemistry Study of the Chemical Bond (1)[J]. 物理化学进展, 2018, 07(01): 9-18. http://dx.doi.org/10.12677/JAPC.2018.71002
参考文献 (References)
- 1. L. 鲍林, 等, 著. 化学键的本质[M]. 卢嘉锡, 等, 译. 上海: 上海科学技术出版社, 1960.
- 2. Parr, R.G., Donnelly, R.A., Levy, N. and Palke, W.E. (1978) Electronegativity: The Density Functional Viewpoint. The Journal of Chemical Physics, 68, 3801. https://doi.org/10.1063/1.436185 Parr, R.G. and Yang, W. (1989) Density-Functional Theory of Atoms and Molecules. Oxford University Press, New York.
- 3. 周光耀. 氢键的量子化学研究(一) [J]. 物理化学进展, 2015, 4(2): 84-101. 周光耀. 氢键的量子化学研究(二) [J].物理化学进展, 2016, 5(2): 58-74.
- 4. Levine, I.N., 著. 量子化学[M]. 宁世光, 余敬曾, 刘尚长, 译. 北京: 人民教育出版社, 1981.
- 5. 杜利利, 王中英, 陈照, 董青松. 金属–金属键化合物的分类及其前景展望[J]. 化学研究, 2016, 27(5): 644-649.
- 6. Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Petersson, G.A., Nakatsuji, H., Li, X., Caricato, M., Marenich, A.V., Bloino, J., Janesko, B.G., Gomperts, R., Mennucci, B., Hratchian, H.P., Ortiz, J.V., Izmaylov, A.F., Sonnenberg, J.L., Williams-Young, D., Ding, F., Lipparini, F., Egidi, F., Goings, J., Peng, B., Petrone, A., Henderson, T., Ranasinghe, D., Zakrzewski, V.G., Gao, J., Rega, N., Zheng, G., Liang, W., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Throssell, K., Montgomery, Jr., J.A., Peralta, J.E., Ogliaro, F., Bearpark, M.J., Heyd, J.J., Brothers, E.N., Kudin, K.N., Staroverov, V.N., Keith, T.A., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A.P., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Millam, J.M., Klene, M., Adamo, C., Cammi, R., Ochterski, J.W., Martin, R.L., Morokuma, K., Farkas, O., Foresman, J.B. and Fox, D.J. (2016) Gaussian 16, Revision A.03. Gaussian, Inc., Wallingford, CT.
- 7. Lu, T. and Chen, F.W. (2012) Multiwfn: A Multifunctional Wavefunction Analyzer. Journal of Computational Chemistry, 33, 580-592. https://doi.org/10.1002/jcc.22885
- 8. Huber, K.P. and Herzberg, G. (1979) Molecular Spectra Molecular Structure IV. Constants of Diatomic Molecules. Van Nostrand Reinhold Company, New York, 104-113. https://doi.org/10.1007/978-1-4757-0961-2
- 9. 庞礼军, 等. H2, HF分子的基态势能函数[J]. 武汉大学学报(理学版), 2007, 53(2): 147-150.
- 10. 李大庆. 我科学家用高分辨原子力显微镜率先“看见”氢键[N/OL]. 光明网, 2013-11-23.
- 11. Hoard, J.L., Geller, S. and Cashin, W.M. (1951) Structures of Molecular Addition Compounds. III. Ammonium-Boron Trifluoride, H3N-BF3. Acta Crystallographica, 4, 396-398. https://doi.org/10.1107/S0365110X5100132X
- 12. Schilt, A.A. (1969) Analytical Applications of 1,10-Phenanthronline and Related Compounds. Pergamon Press, London.
- 13. Ghosh, D.C. and Bhattacharyya, S. (2010) Computation of Quantum Mechanical Hybridization and Dipole Correlation of the Electronic Structure of the F3B-NH3 Supermolecule. International Journal of Quantum Chemistry, 105, 270-279. https://doi.org/10.1002/qua.20690
- 14. K. 彼得, 等, 著. 有机化学: 结构与功能[M]. 戴立信, 等, 译. 第4版. 北京: 化学工业出版社, 2006.