Asian Case Reports in Pediatrics
Vol.06 No.03(2018), Article ID:26297,8
pages
10.12677/ACRP.2018.63005
A Child with Geleophysic Dysplasia Type 2 Caused by a Novel Mutation of FBN1: A Case Report and Literature Review
Yang Li, Huahong Wu, Hui Li
Department of Growth and Development, Capital Institution of Pediatrics, Beijing

Received: Jul. 14th, 2018; accepted: Jul. 31st, 2018; published: Aug. 8th, 2018

ABSTRACT
Objective: To report a geleophysic dysplasia type 2 (GD2) caused by FBN1 gene mutation, provide the clinical features of early diagnosis in patients with GD. Method Analysis was performed on clinical manifestation, imageological examination and parental Sanger test of a patient carrying novel mutation of FBN1, and literatures about clinical features of GD. Results: A 10-year-and-11-month-old boy referred to the growth and development clinic for short stature. Major clinical manifestations are short stature, special face, heart valvular disease, digits joint stiffness and skeletal dysplasia. A pair of novel heterozygous mutation of FBN1 was found by gene sequencing. Nucleotide change is c.5284G>A and amino acid change is p.G1762S (Gly to Ser). Databases were searched including Pubmed Database, Wan Fang Database, CNKI Database, OMIM Database from database-building time to December 1, 2017 by the use of key words “geleophysic dysplasia” and “FBN1”. A total of 23 articles were retrieved and 6 English articles (32 cases) were screened out, including three familial cases. Conclusion: Geleophysic dysplasia (GD) is a rare skeletal disorder, which is liable to cause delayed diagnose and misdiagnosis. Careful examination is recommended for patients with heterogeneity short stature, short hand and feet, special face, heart valvular disease and joint stiffness. We should provide multidisciplinary monitoring to GD patients, like ultrasonic cardiogram, abdominal ultrasound, pulmonary function and so on.
Keywords:Geleophysic Dysplasia Type 2, FBN1 Gene, Short Stature
FBN1基因突变导致Geleophysic发育不良2型1例并文献复习
李阳,武华红,李辉
首都儿科研究所生长发育研究室,北京
收稿日期:2018年7月14日;录用日期:2018年7月31日;发布日期:2018年8月8日

摘 要
目的:报告1例FBN1基因新生突变所致的Geleophysic发育不良2型(GD2),探讨GD临床诊治特点,为GD患儿的早期发现、早期诊断提供依据。方法:总结患儿临床特征表型、实验室检查、基因测序结果,并对FBN1基因突变导致的GD病例进行文献复习。结果:本例患儿男,10岁11月,因“身材矮小”初诊,主要表现为身材矮小、特殊面容、心脏瓣膜病、手指关节僵硬和骨骼发育不良。遗传病综合基因测序发现FBN1新生基因突变,核苷酸改变为c.5284G>A,氨基酸改变为p.Gly1762Ser。检索Pubmed数据库、万方数据库、中国知网、OMIM数据库,检索时间从建库至2017年12月,共检索到23篇文献,其中筛选出6篇英文文献共有32例由FBN1基因导致的GD病例报道,与本文1例合并后共33例。对该病的病因、临床表型、诊断和治疗随访进行了文献综述。结论:GD在临床罕见,极易误诊和漏诊,身材矮小合并短手短脚、心脏瓣膜病及关节僵硬时,应高度警惕GD的可能性。诊断GD后对患儿需长期多学科监测随访。
关键词 :Geleophysic发育不良2型(GD2),FBN1基因,身材矮小

Copyright © 2018 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 引言
肢端发育不良是一组以身材矮小、短手短脚、关节僵硬、皮肤增厚和骨骼畸形等为主要特征的疾病 [1]。2015年修订的遗传性骨病疾病分类中,肢端发育不良包括10种疾病,其中FBN1基因突变导致的疾病有Geleophysic发育不良(GD)、Acromicric发育不良(AD)、Weill-Marchesani综合征(WMS) [2]。尽管这些疾病在外貌特征上相似,但是每种疾病有其特有表现。GD有特有的心脏瓣膜病变、呼吸功能不全、肝脏肿大甚至有早期死亡的可能;AD特有的声音嘶哑;WMS有特有的晶状体半脱位、白内障和球状晶状体等 [1]。GD是由Vanace等 [3]于1960年首次报告,Spranger J等 [4]1971年将该病命名为GD。GD可由ADMTSL2 (MIM 231050)和FBN1 (MIM 614185)两种基因突变导致,绝大多数GD由ADMTSL2突变导致,称为GD1型。Le Goff等 [5]2011年首次提出FBN1突变导致的GD称为GD2型。GD2型特有的心脏瓣膜病变通常为渐进性改变,可能导致继发性心肌肥大、心脏衰竭等。本文报道中国大陆地区1例FBN1基因c.5284G>A位点突变导致的GD患者,并进行文献综述,旨在提高临床医生对GD2型的认识和诊治水平,有利于提高患儿的生存率和生活质量。
2. 资料与方法
2.1. 一般资料
该病例报告已获得患儿家属知情同意。患儿男,系第2胎第2产,生育间隔1年3月,孕足月、顺产,出生身长和出生体重不详,出生后无产伤、窒息。母亲孕期无妊娠并发症,否认特殊用药及化学性物质接触史。患儿出生后即生长缓慢,2岁时发现患有先天性主动脉狭窄,未予外科干预治疗。10岁11月时因“身材矮小”于2017年7月21日于我院生长发育门诊就诊。日常饮食可,无挑食偏食,运动及睡眠可。父亲身高168 cm,母亲身高155 cm,哥哥身高正常(具体数据不详)。患儿父母否认近亲结婚,无家族性遗传病史,家族成员无类似病史。临床体征见图1。
体格检查:身高112.4 cm (−5.01SD) (132.1~158.9 cm),体重26.2 kg (−1.83SD) (25.64~57.58kg) [6],坐高69.3 cm (−2.06SD) (69.6~84.4cm) [7],BMI 20.74 kg/m2 (+1.15SD) (13.73~24.17 kg/m2) [8];特殊面容:面部饱满、鼻短、塌鼻梁、人中长,短手短脚、手指短、指间关节挛缩(不影响日常活动)、四肢长骨短、皮肤增厚等特殊躯体特征;智力发育正常;心音有力,节律整齐,心前区可闻及III级收缩期杂音,肺腹部查体未见异常,甲状腺未扪及肿大。一般实验室检查:甲功:TT3 2.22 nmol/L、FT 5.93 pmol/L、TT4 123 nmol、FT4 17.3 pmol/L、TSH 2.6249 mIU/L。IGF-1:150 ng/ml。GH 运动前1.8 ng/mL,运动后5.89 ng/mL。其余检查(生化等)均未见明显异常。
影像学:1) 超声心动图:主动脉瓣狭窄(中度)伴少量返流。2) 腹部超声:肝胆胰脾肾未见明显异常。3) 2017年7月6日左手正位骨龄片:骨龄8.5岁,落后于实际年龄2.5岁。骨龄片见骨端发育不良、掌骨形态不规则;第2掌骨左侧不规则凹陷及第5掌骨右侧不规则凹陷。2013年7月18日(年龄:6岁11月)左手正位骨龄片:骨龄5.5岁,落后于实际年龄1.4岁。4) 脊柱及骨盆X线正侧位片:可见髋臼和股骨头发育不良,脊柱弯曲度及脊柱间隙未见异常。
基因检测:临床诊断“软骨发育不全”,征得患儿家长知情同意,采集患儿及父母的外周血样本,用二代测序方法对患儿进行遗传病综合检测,并结合Sanger测序验证。基因测序及验证结果(见表1和图2),患儿检出FBN1新生突变,患儿父母未检出该变异。根据基因序列突变诊断国际规范 [9],该突变位于GD2型的主要突变位点42号外显子区域且为已报道的致病性突变,故诊断为GD2型。
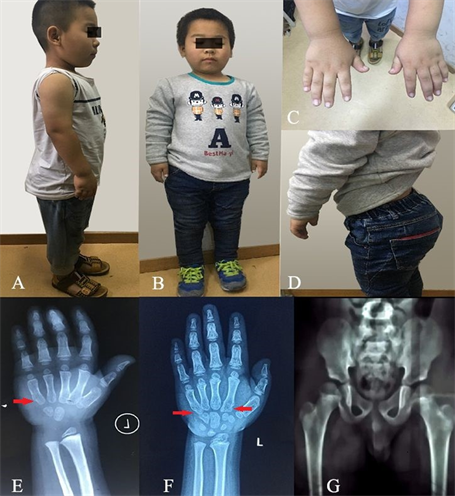
Figure 1. Clinical characteristics of the patient. (A, B): short stature and shortened long tubular bone; C: short hand and joint contracture; D: increased lumbar lordosis; (E, F): delayed bone age in 2013 and 2017; G: X-ray film of pelvis and femur
图1. 患儿临床特征图。(A、B):矮身材和长骨变短;C:短手、指关节挛缩;D:腰椎前凸增加;(E、F):2013年和2017年骨龄明显落后;G:骨盆及股骨正位X线片

Table 1. Gene sequencing results
表1. 患儿基因测序结果

Figure 2. Amionoacid change and Sanger validation results
图2. 患儿氨基酸改变及Sanger验证结果
2.2. 方法
以“geleophysic dysplasia”、 “geleophysic发育不良”以及“FBN1”为检索词,分别检索建库至2017年12月1日前的Pubmed数据库、万方数据库、CNKI数据库、OMIM数据库。共检索出23篇英文文献,筛选出其中6篇英文文献报道32个病例由FBN1突变导致的GD [5][10][11][12][13][14],其中国内8个病例,国外24个病例,有3个家族性报道,其中有13例有详细的病例资料。33个病例基因改变及GD特征性改变总结见表2,14例有详细病例资料面部及骨骼畸形特征出现频率统计见表3。
3. 讨论
GD是罕见的由基因异常导致的骨骼发育不良,临床主要表现为:身材矮小、短手短脚、关节活动受限、皮肤增厚、面部特征和心脏瓣膜病变、肝肿大、呼吸功能不全等器官病变 [1]。目前以致病基因不同分为两型,GD1由ADMTSL2突变引起为隐性遗传,GD2由FBN1突变引起为显性遗传,两者在临床特征上未见明显区别。该病在人群中发病率不详,GD1患者的每个同胞兄妹有25%的机率成为患者,50%的机率成为无临床表现的携带者,25%的机率成为正常人且非携带者。GD2如父母双方有一人为患者则子代50%的机率成为患者,如果父母双方无人患有GD2,则子代可能因为生殖系嵌合有1%~2%的可能性患有GD2 [15]。
GD2致病基因FBN1定位于15q21.1,全长 > 600 kb,其cDNA长度约为10 kb,包含65个编码外显子,编码原纤维蛋白1 (Fibrillin-1)。Fibrillin-1共包含6个结构域,其中主要结构域有① 47个类上皮生长因子样结构域(EGF-like-domain),该结构域包含6个半胱氨酸残基,形成3对二硫键。47个结构域中有43个是高度保守一致序列,它们与钙结合称为钙结合-类上皮生长因子结构域(cbEGF-like-domain),保护
Table 2. FBN1 gene changes and clinical manifestations of 33 GD2 patients
表2. 33例FBN1基因改变及特征性改变总结
fibrillin-1的杆状结构,使类上皮生长因子样结构域之间连接稳固,免于fibrillin-1被蛋白酶水解 [16]。若钙离子的结合能力降低,暴露隐藏的蛋白酶切位点,容易导致蛋白质产物的结构不稳定 [17]。② 9个转化生长因子β结合结构域(transforming growth factor-β binding domain TB domain),是细胞外基质微纤维结构主要组成部分,主要参与细胞的生长和维持。主要表达在血管、软骨、肾脏、肾脏、皮肤、尿道、骨细胞、
Table 3. Facial and skeletal features of 14 GD2 patients
表3. 14例有详细病例资料面部及骨骼畸形特征统计
眼球晶状体悬韧带。③ 8-半胱氨酸结构域(8-cys domain)。迄今为止的报道中FBN1突变导致GD的突变位点多集中在41~42号外显子区域,编码TB5 (fifth TGF-β binding domain)结构域 [5],本例患者FBN1突变(p.Gly1762Ser)位于42号外显子区域。Le Goff等认为TB5结构域和矮身材、短手及关节僵硬有关,可能原因是FBN1基因突变使TGF-β的调节失调导致TGF-β释放失控含量增加并且增强TGF-β活性,诱发身材矮小、短手等表型出现。8-半胱氨酸通过蛋白质链内二硫键连接直接参与FBN1折叠,TB5蛋白区域突变有50%的原因是因为半胱氨酸缺失或生成增加,影响二硫键的形成,进而影响蛋白质结构 [5]。Mclnerney-Leo AM等 [18]最新研究发现LTBP3 (transforming growth factor-β binding protein-3)基因杂合突变也和GD有关,LTBPs和FBNs有较高的同源性,两者都有钙结合-类上皮生长因子结构域和8-半胱氨酸结构域,而且8-半胱氨酸结构域仅FBN和LTBP蛋白质家族共有 [19]。
FBN1突变一方面可导致身材矮小(肢端发育不全)的疾病,另一方面可导致身材高大(马凡综合征)的疾病。FBN1突变发生两种相反临床表型的原因,基因型和表型的关系尚不清楚。1991年Dietz等 [20]首次提出FBN1突变是马凡综合征的致病原因,FBN1突变导致马凡综合征主要突变位点在13、15、24~28、32和43号外显子区域 [21]。
GD目前诊断主要依靠临床特征及影像学检查,确诊依靠基因外显子测序。临床特征主要有:严重身材矮小、短手短脚、进行性关节僵硬、皮肤增厚、心脏瓣膜病变。特征性面部特征有:圆脸、小鼻伴鼻梁宽、上嘴唇薄、长或平人中、眼睑狭窄、眼距宽。除此之外还有肝肿大、气管狭窄、呼吸功能不全和反复中耳炎 [15]。如表3所示14例有详细病例资料的病人特征描述中面部特征最常见的表型为宽鼻梁和圆脸,其次为眼距宽、长人中、肉鼻,最后为长嘴唇薄、小鼻及眼睑狭窄。影像学表现主要有:明显骨龄落后、近节指骨缩短、锥形骨骺、长管骨缩短和小骨附生。
对于FBN1突变导致的GD目前没有合适有效的治疗方法,有2例病人由于确诊时间晚,早期被诊断为特发性矮身材,根据标准的儿科内分泌诊疗指南进行重组人生长激素治疗,这2例患者在治疗过程中没有显著的追赶生长 [12]。Le Goff等 [5]认为GD的心脏瓣膜病变呈渐进性发展,需要长期监测超声心动图对心脏情况进行随访观察。在现有的对GD进行随访的病例报告中,Titomanlio L等 [22]对一患病男孩进行7年随访心脏异常在前期(9月龄~4岁)时心脏功能降低,但后期(4岁~7岁)时心功能未见变化,相反肺动脉瓣狭窄较前改善。Scott A等 [23]对两位患者进行随访发现,男孩(0月龄~14岁)心功能逐渐降低需要外科手术进行干预,而女孩(2月龄~7岁)心功能未见明显变化。Elhoury ME等 [24]对三位因ADMTSL2突变导致的GD1进行1~2年随访观察发现三位患儿的心功能较前均无明显改变。这提示GD患儿心脏瓣膜病致心功能变化可能与患病时间长短、性别及基因突变类型有关。有文献报道关节受限和肝脏肿大呈渐进性改变 [13],所以有必要对GD患儿需要长期跟踪随访超声心动图、腹部超声、关节功能。严重GD可因气管狭窄、支气管肺功能不全而早期死亡 [5],因此多学科长期检测对于提高患儿的生命质量十分重要。
4. 结论
总之,GD2在临床上罕见,且与软骨发育不全等疾病难以鉴别,极易延误诊断或漏诊,并且因患者存在的显性遗传特性以及进行性的心脏、关节及内脏病变,对患者及子代的生存和生活质量有重大影响。提高对本病的诊治水平和诊断率,可早期干预改善患者生存质量,提高长期生存率及降低子代患病风险。本文通过报道一例我院最新诊断的GD2患儿和相关的文献综述提示,当患儿有严重身材矮小,合并特殊面容、短手短脚、心脏瓣膜病及关节僵硬等临床特征时,应高度警惕GD的可能性。
文章引用
李 阳,武华红,李 辉. FBN1基因突变导致Geleophysic发育不良2型1例并文献复习
A Child with Geleophysic Dysplasia Type 2 Caused by a Novel Mutation of FBN1: A Case Report and Literature Review[J]. 亚洲儿科病例研究, 2018, 06(03): 27-34. https://doi.org/10.12677/ACRP.2018.63005
参考文献
- 1. Le, G.C. and Cormier-Daire, V. (2012) From Tall to Short: The Role of TGFβ Signaling in Growth and Its Disorders. American Journal of Medical Genetics Part C Seminars in Medical Genetics, 160C, 145-153.
https://doi.org/10.1002/ajmg.c.31337 - 2. Bonafe, L., Cormier-Daire, V., Hall, C., et al. (2011) Nosology and Classification of Genetic Skeletal Disorders: 2015 Revision. American Journal of Medical Genetics Part A, 167A, 2869-2892.
- 3. Vanace, P.W., Friedman, S. and Wagner, B.M. (1960) Mitral Stenosis in an Atypical Case of Gar-goylism: A Case Report with Pathologic and Histochemical Studies of the Cardiac Tissues. Circulation, 21, 80-89.
- 4. Spranger, J.W., Gilbert, E.F., Tuffli, G.A., et al. (1971) Geleophysic Dwarfism—A “Focal” Mucopoly-saccharidosis? Lancet, 2, 97-98.
https://doi.org/10.1016/S0140-6736(71)92073-3 - 5. Goff, C.L., Mahaut, C., Wang, L.W., et al. (2011) Mutations in the TGFβ Binding-Protein-Like Domain 5 of FBN1, Are Responsible for Acromicric and Geleophysic Dysplasias. American Journal of Human Genetics, 89, 7-14.
https://doi.org/10.1016/j.ajhg.2011.05.012 - 6. 李辉, 季成叶, 宗心南, 等. 中国0-18岁儿童、青少年身高、体重的标准化生长曲线[J]. 中华儿科杂志, 2009, 47(7): 487-492.
- 7. 张亚钦, 李辉, 季成叶. 中国0至18岁正常儿童青少年身体比例生长规律的研究[J]. 中国循证儿科杂志, 2010, 5(5): 349-354.
- 8. Zong, X.N. and Li, H. (2013) Construction of a New Growth References for China Based on Urban Chinese Children: Comparison with the WHO Growth Standards. PLoS ONE, 8, e59569.
https://doi.org/10.1371/journal.pone.0059569 - 9. Sue, R., Nazneen, A., Sherri, B., et al. (2015) Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Mo-lecular Pathology. Genetics in Medicine, 17, 405-424.
https://doi.org/10.1038/gim.2015.30 - 10. Cheng, S.W., Luk, H.M., Chu, Y.W.Y., et al. (2018) A Report of Three Families with FBN1-Related Acromelic Dysplasias and Review of Literature for Genotype-Phenotype Correlation in Gelophysic Dysplasia. European Journal of Medical Genetics, 61, 219-224.
https://doi.org/10.1016/j.ejmg.2017.11.018 - 11. Hasegawa, K., Numakura, C., Tanaka, H., et al. (2016) Three Cases of Japanese Acromicric/Geleophysic Dysplasia with FBN1 Mutations: A Comparison of Clinical and Radiological Features. Journal of Pediatric Endocrinology and Metabolism, 30, 117-121.
- 12. Bruin, C.D., Finlayson, C., Funari, M.F.A., et al. (2016) Two Patients with Severe Short Stature Due to a FBN1 Mutation (p.Ala1728Val) with a Mild Form of Acromicric Dysplasia. Hormone Research in Paediatrics, 86, 342-348.
https://doi.org/10.1159/000446476 - 13. Wang, Y., Zhang, H., Ye, J., et al. (2014) Three Novel Mutations of the FBN1 Gene in Chinese Children with Acromelic Dysplasia. Journal of Human Genetics, 59, 563-567.
https://doi.org/10.1038/jhg.2014.73 - 14. Lee, T., Takeshima, Y., Okizuka, Y., et al. (2013) A Japanese Child with Geleophysic Dysplasia Caused by a Novel Mutation of FBN1. Gene, 512, 456-459.
https://doi.org/10.1016/j.gene.2012.10.060 - 15. Adam, M.P., Ardinger, H.H., Pagon, R.A., et al. (2016) Gene Reviews. University of Washington, Seattle.
- 16. Whiteman, P. and Handford, P.A. (2003) Defective Secretion of Recombinant Fragments of Fibrillin-1: Implications of Protein Misfolding for the Pathogenesis of Marfan Syndrome and Related Disorders. Human Molecular Genetics, 12, 727-737.
https://doi.org/10.1093/hmg/ddg081 - 17. 邓婷, 李杨. 先天性晶状体脱位与FBN1基因[J]. 中华实验眼科杂志, 2008, 26(2): 157-160.
- 18. Mcinerney-Leo, A.M., Le Goff, C., Leo, P.J., et al. (2016) Mutations in LTBP3 Cause Acromicric Dysplasia and Geleophysic Dysplasia. Journal of Medical Genetics, 53, 457-464.
https://doi.org/10.1136/jmedgenet-2015-103647 - 19. Robertson, I., Jensen, S. and Handford, P. (2011) TB Domain Proteins: Evolutionary Insights into the Multifaceted Roles of Fibrillins and LTBPs. Biochemical Journal, 433, 263-276.
https://doi.org/10.1042/BJ20101320 - 20. Dietz, H.C., Cutting, C.R., Pyeritz, R.E., et al. (1991) Marfan Syndrome Caused by a Recurrent de Novo Missense Mutation, in the Fibrillin Gene. Nature, 352, 337-339.
https://doi.org/10.1038/352337a0 - 21. Collod-Béroud, G., Le, B.S., Ades, L., et al. (2003) Update of the UMD-FBN1 Mutation Database and Creation of an FBN1 Polymorphism Database. Human Mutation, 22, 199-208.
https://doi.org/10.1002/humu.10249 - 22. Titomanlio, L., Della, C.R., Lecora, M., et al. (1999) Geleophysic Dysplasia: 7-Year Follow-Up Study of a Patient with an Intermediate Form. American Journal of Medical Genetics, 86, 82-85.
https://doi.org/10.1002/(SICI)1096-8628(19990903)86:1<82::AID-AJMG16>3.0.CO;2-O - 23. Scott, A., Yeung, S., Dickinson, D.F., et al. (2005) Natural History of Cardiac Involvement in Geleophysic Dysplasia. American Journal of Medical Genetics Part A, 132A, 320-323.
https://doi.org/10.1002/ajmg.a.30450 - 24. Elhoury, M.E., Faqeih, E., Almoukirish, A.S., et al. (2015) Cardiac Involvement in Geleophysic Dysplasia in Three Siblings of a Saudi Family. Cardiology in the Young, 25, 81-86.
https://doi.org/10.1017/S1047951113001753