Advances in Clinical Medicine
Vol.
13
No.
03
(
2023
), Article ID:
63246
,
7
pages
10.12677/ACM.2023.133676
脊髓小脑共济失调2型1家系报道
王文1,张璇2*
1济宁医学院临床医学院,山东 济宁
2济宁医学院附属济宁市第一人民医院神经内科,山东 济宁
收稿日期:2023年2月24日;录用日期:2023年3月19日;发布日期:2023年3月29日

摘要
通过对脊髓小脑共济失调2型1家系报道分析,总结其发病特点、临床表现、遗传情况及治疗方案并对相关文献进行复习。
关键词
脊髓小脑共济失调,系谱,病例报告

Spinocerebellar Ataxia Type 2: A Family Report
Wen Wang1, Xuan Zhang2*
1Clinical Medical College, Jining Medical University, Jining Shandong
2Neurology Department, Jining First People’s Hospital Affiliated to Jining Medical University, Jining Shandong
Received: Feb. 24th, 2023; accepted: Mar. 19th, 2023; published: Mar. 29th, 2023

ABSTRACT
Through the analysis of the report of a family with spinocerebellar ataxia type 2, this paper summarized its characteristics, clinical manifestations, heredity and treatment plan, and reviewed the relevant literature.
Keywords:Spinocerebellar Ataxia, Pedigree, Case Reports

Copyright © 2023 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/


1. 引言
脊髓小脑共济失调2型(spinocerebellar ataxia 2, SCA2)是一种常染色体显性遗传性小脑性共济失调,以进行性小脑综合征为特征,伴有扫视减慢、周围神经病变、认知障碍和其他多系统特征 [1],临床上多数患者表现为共济失调、构音障碍、腱反射减弱或消失,也有表现为帕金森样症状和认知下降,但其少见且容易误诊。现报道我科收治的脊髓小脑共济失调2型1家系。
2. 临床资料
先证者(IV8)女,38岁,因“行走不稳、双手抖动1年余”于2022年01月14日来我院门诊就诊。患者1年余前无明显诱因出现行走不稳,主要为向后倾斜感,双手不自主抖动,伴言语不清,表现为吐词含糊不清,伴吞咽困难,伴饮水呛咳,无视物模糊、视物旋转,无肢体无力,无意识障碍。患者既往体健,否认高血压病、糖尿病、冠心病等病史,否认肝炎、结核等传染病病史及密切接触史,否认外伤、手术史,否认输血史,否认食物、药物等过敏史。
家族史:1) 先证者曾外祖母(I1):发现有行走不稳及肢体抖动症状,去世较早。2) 先证者外祖父(II5):患者诉与其有类似症状,且去世较早。3) 先证者外祖父的哥哥(II2):曾发现行走不稳及肢体抖动,已去世。4) 先证者母亲(III9):37岁时发现行走不稳,吞咽困难,发病5~6年时生活尚可自理,病情逐渐加重,19年后完全卧床,行动不能,59岁时去世。5) 先证者母亲的二表姐(III2):30多岁时发现行走不稳,病情逐渐加重,48岁时去世。6) 先证者母亲的三表姐(III4):40岁时发现双手抖动,易摔倒,病情逐渐加重,现生活不能自理,现年60岁,完善基因测序如下图(见图1)。7) 先证者母亲的五表妹(III7):发现行走迟缓及言语低沉,已去世,且去世较早。8) 先证者弟弟(IV9):现年38岁,16岁时发现右手不自主抖动,活动后加重,持物时明显,饮酒后抖动可缓解,症状未见明显加重。既往体健。查体:意识清楚,精神可。言语清楚流利,四肢肌力5级,右侧肢体肌张力稍增高,左侧肢体肌张力正常。腱反射正常,病理征未引出,双侧指鼻试验欠稳准,双侧跟膝胫试验尚稳准,轮替试验正常。汉密尔顿抑郁评分(HAMD) 0分,汉密尔顿焦虑评分(HAMA) 2分。完善甲功五项未见明显异常。9) 先证者大表姐(IV1):34岁时发现双手抖动及动作迟缓,病情逐渐加重,已去世且去世较早。10) 先证者二表姐(IV2):现年39岁,35岁时发现双下肢行动迟缓,左上肢为著,行走无力,启步稍困难,伴左手不自主抖动,伴饮水呛咳,伴言语不清,主要为吐词含糊不清,生活自理能力差。既往2型糖尿病病史。查体:意识清楚,精神可。面具脸,言语欠清晰流利,左上肢静止性震颤,右上肢姿势性震颤,四肢肌力5级,四肢肢体肌张力增高,以双下肢为著。腱反射正常,病理征未引出,双侧指鼻试验欠稳准,双侧跟膝胫试验欠稳准,轮替试验缓慢。简易智力状态检查(MMSE)评分29分,MoCA评分28分(文化程度:初中)。4年前前来我院就诊,完善相关辅助检查:血常规、尿常规、粪便常规加隐血、肝功、肾功、电解质、凝血五项、同型半胱氨酸、癌抗原CA-125均大致正常。空腹血糖:8.55 mmol/L,糖化血红蛋白:10.8%。完善遗传病基因分析报告提示发现ATXN2基因的1个变异,关联疾病为:迟发型帕金森病(易感) (OMIM: 168600) ;发现EIF4G1基因的1个变异,关联疾病为:帕金森病18型(OMIM: 614251),基因测序如下图(见图2)。给予多巴丝肼0.375 g/d,分三次服用;普拉克索0.5 mg/d,分两次服用;复方卡比多巴一片半,分三次服用;文拉法辛缓释胶囊早75 mg。于我院复诊,患者诉行动迟缓及言语不清好转。11) 先证者三表妹(IV5):出现类似行走不稳及肢体抖动症状,其本人拒绝相关询问及查体。
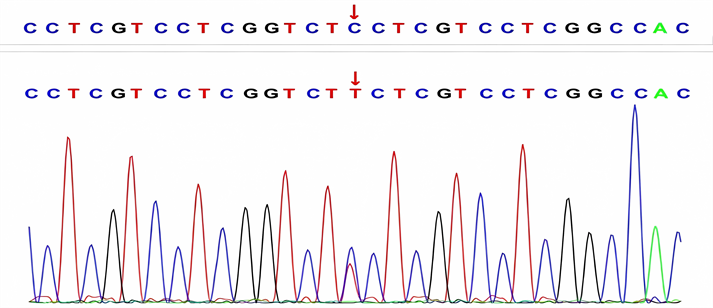
Figure 1. Gene sequencing results of III4 in the family diagram
图1. 家系图中III4的基因测序结果
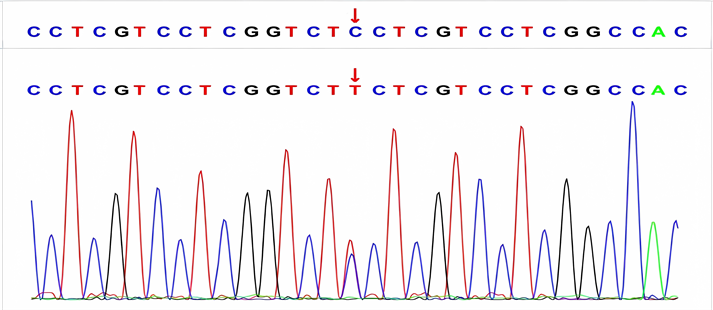
Figure 2. Gene sequencing results of IV2 in the family diagram
图2. 家系图中IV2的基因测序结果
入院查体:心、肺、腹部查体无特殊。神经系统查体:意识清楚,精神可,吟诗样语言,记忆力、理解力、定向力、计算力粗测正常。双侧瞳孔等大等圆,直径约3 mm,对光反射灵敏,双眼眼球活动灵活,未见复试及眼震。双侧额纹、鼻唇沟对称,伸舌居中,余颅神经大致正常。四肢肌力、肌张力正常。双侧腱反射未引出,深浅感觉检查未见明显异常。双侧病理征阴性。脑膜刺激征阴性。双侧指鼻、跟膝胫试验大致稳准,直线行走试验阳性。全身肌肉无萎缩。
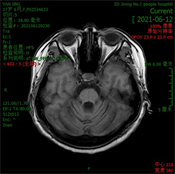
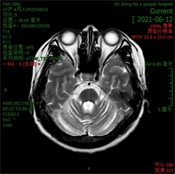
辅助检查:血常规、尿常规、粪便常规及隐血大致正常。肝功能、肾功能、血脂未见明显异常,血糖12.74 mmol/L。甲状腺功能五项正常,贫血三项未见异常。简易智力状态检查(MMSE)评分29分,MoCA评分27分(文化程度:初中)。MRI (见图3):双侧大脑半球结构对称,脑实质内未见异常信号灶,DWI (b = 1000)未见明显异常弥散受限高信号影;小脑脑沟增宽,第四脑室增宽,余各脑室、脑池、脑裂及脑沟大小、形态正常,中线结构居中。患者因自身原因拒绝微量元素等其他检查。
 (a)
(a)
 (b)
(b)
 (c)
(c)
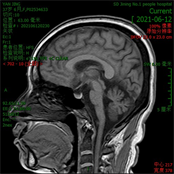 (d)注:(a) T1WI;(b) T2WI;(c) FLAIR;(d) T1WI矢状位。
(d)注:(a) T1WI;(b) T2WI;(c) FLAIR;(d) T1WI矢状位。
Figure 3. Brain MRI results of patients
图3. 患者的颅脑MRI结果
基因检测结果:在依据“知情同意原则”并充分保护患者隐私的前提下,空腹采集患者外周静脉血行共济失调相关10种动态突变检测,运用荧光PCR-毛细管电泳法,结果提示患者ATXN2基因两条等位基因三碱基重复区域(CAG) n重复次数为20/38,SCA2的患病风险较高(见图4),trio全外显子组测序检测未发现疾病相关性较高的变异。

Figure 4. Gene testing results of the patient (proband) (fluorescence PCR-capillary electrophoresis)
图4. 患者(先证者)的基因检测结果(荧光PCR-毛细管电泳法)
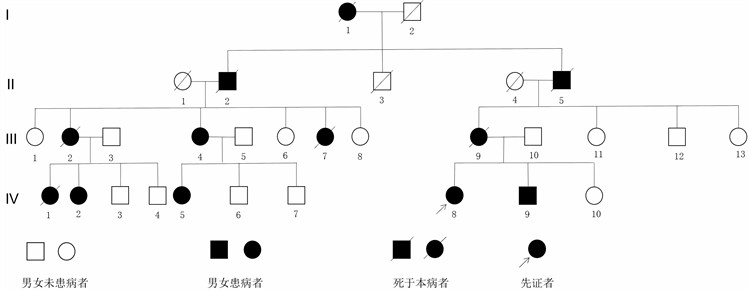
Figure 5. Genetic family diagram of a patient with spinocerebellar ataxia type 2, with IV8 as the patient
图5. 脊髓小脑共济失调2型患者遗传家系图,IV8为本例患者
遗传方式:家系连续遗传4代,患病者12例,男女比例为3:9,观察家系图提示符合常染色体显性遗传,该家系遗传图谱见图5。
3. 讨论
遗传性共济失调是一种具有遗传异质性的神经退行性疾病,由小脑及其传入和传出连接、脊髓、周围神经和脑干退行性变引起 [1]。而脊髓小脑性共济失调(spinocerebellar ataxia, SCA)是最常见的类型,其中7种(SCA1, SCA2, SCA3, SCA6, SCA7, SCA17, DRPLA)是由突变基因编码区病理性胞嘧啶–腺嘌呤–鸟嘌呤(CAG)三核苷酸重复扩增引起的 [2] [3] [4]。SCA2是仅次于SCA3的全球第二大流行的亚型 [5]。本病最先由Wadia和Swami在印度发现,二十多年后,在古巴发现更多的呈现家族性遗传的患者 [6],而在澳大利亚、突尼斯、德国、意大利、墨西哥、波兰等国家也发现存在不少家族病例 [7] [8]。从遗传学上讲,SCA2是由定位于染色体12q24.1 [9] 的ATXN2基因编码区CAG DNA重复扩增引起的,因此在ataxin-2蛋白中表达有毒的多聚谷氨酰胺扩增,从而导致小脑浦肯野细胞和其他细胞中的几个桥脑、中脑和丘脑神经元的进行性神经元死亡 [1]。SCA2的潜在突变包括位于ATXN2基因第一外显子内的翻译的三核苷酸CAG重复序列的扩展 [10] [11] [12]。在正常个体中,等位基因包含13到31个CAG重复,有22个CAG重复的未展开等位基因频率最高(76%),其余正常等位基因中,5%的范围在13到21个CAG单位之间,19%的范围在23到31个单位之间 [13]。SCA2病理等位基因有超过32个CAG重复,但在32~36个范围内,它们具有不完全外显 [11] [14] [15]。在该家系遗传中,男女均可发病,且性别遗传无关,符合常染色体显性遗传。SCA2发病年龄多在30~40岁,存在遗传早现现象 [16],该家系发病者多在此区间,也存在遗传早现现象,先证者母亲于38岁发病,先证者37岁发病,而先证者弟弟于17岁发病。共济失调评定量表(SARA)显示,SCA2患者的病程越长,病情越严重,平衡能力越差,功能依赖性越强 [17]。
神经病理学分析显示SCA2患者小脑、基底神经节和额叶广泛萎缩 [18] [19]。最近对皮层灰质和白质结构复杂性指标的分形分析表明,SCA2患者体内小脑和大脑皮层的结构复杂性降低 [20]。在SCA2患者中,MRI显示橄榄脑桥小脑萎缩,临床发作前桥脑脑干体积减少,桥脑弥漫性自旋–自旋松弛时间(T2)高信号,小脑脚中部和小脑白质呈“热交叉”征,晚期丘脑和顶叶皮质萎缩,而就脑桥脑干和小脑的体积而言,脑干和小脑的弥散张量成像指数是监测SCA2疾病进展的最有前景的影像指标 [21] [22]。最近的MRI研究显示SCA2患者的下橄榄核肥大,这是SCA2与其他遗传性SCAs相比的一个独特特征 [23]。先证者的颅脑MRI提示小脑萎缩,因其发病时间较短,影像学表现相对较轻。影像学表现可以在一定程度上辅助本病的诊断。
所有SCA2均表现为进行性小脑综合征,以共济失调步态、小脑构音障碍、运动障碍、扫视变慢为特征 [1],此外,还有一些临床症状表现为周围神经病变、吞咽异常、自主神经功能异常、睡眠障碍、认知障碍 [1] [12] [24],但在某些情况下,帕金森病也被报道为SCA2的临床特征 [25],例如,一个长期(长达34年)的无小脑性共济失调的左旋多巴反应性帕金森综合征的韩国SCA2家系 [26]。也有报告发现SCA2突变的临床表现为多巴胺反应性不对称帕金森病(PD)表型,在日本和中国可能更常见,在这些国家,SCA2被确认为家族性晚发性帕金森病的原因,有高达10%的家庭 [27] [28] [29] [30]。在没有共济失调或扫视减慢的情况下,一些患者还表现为静止性震颤、僵直、运动迟缓,对左旋多巴反应良好,并发现SCA2基因突变较小 [31]。先证者家系存在两种不同的临床表现。IV2患者主要表现为帕金森综合征,表现为左上肢震颤、行动迟缓,且服用左旋多巴后症状改善明显,患者完善基因分析报告提示可能为迟发型帕金森,追问患者家族史,其母亲及姨母多与患者表现大致相同。而先证者主要表现为小脑性共济失调,以共济失调步态、构音障碍、吞咽障碍为主要表现,先证者的基因分析报告提示CAG重复次数为20/38,SCA2患病风险高。给予左旋多巴治疗后,患者症状改善不明显。根据患者临床表现、家族史及辅助检查可以考虑诊断为SCA2。本家系中患者以共济失调步态、构音障碍、运动障碍、吞咽障碍为主要临床表现,扫视变慢、周围神经病变、自主神经功能异常、认知障碍则表现的不明显,考虑来就诊患者发病时间短,而发病时间相对较长的患者未能前来就诊。
SCA2的诊断主要依靠基因测序,其他方法无法明确诊断。治疗上目前尚无根治方法,主要治疗手段为物理治疗和康复治疗,SCA2神经康复治疗的目标是改善协调性、体位功能、行走距离、减少跌倒、改善步态稳定性、更正常的步态速度、步长和节奏以及增加日常生活活动的独立性 [1]。有实验研究发现,经过3个月的日常康复后,68%的康复受试者在协调能力、姿势稳定性、眼运动能力和语言能力方面有显著改善 [32],75%的患者在记忆、注意力和额叶执行能力等认知功能方面有部分恢复 [32]。对于SCA2的治疗,有文献提出两种治疗方案,一种是开发ATXN2的已知功能作为SCA2的治疗靶点,包括谷氨酸信号、钙稳态和RNA代谢,另一种有前途的方法是直接靶向调控ATXN2的表达 [33]。但是这些治疗方案仍处于实验阶段,长期疗效及安全性尚未可知。
致谢
感谢患者提供相关病情资料,国家科学基金对本文的支持及导师对论文写作的指导。
基金项目
国家重点研发计划项目(2018YFC1312001);国家重点研发计划项目(2016YFC0105900)。
文章引用
王 文,张 璇. 脊髓小脑共济失调2型1家系报道
Spinocerebellar Ataxia Type 2: A Family Re-port[J]. 临床医学进展, 2023, 13(03): 4719-4725. https://doi.org/10.12677/ACM.2023.133676
参考文献
- 1. Velazquez-Perez, L., Rodriguez-Labrada, R., Garcia-Rodriguez, J.C., et al. (2011) A Comprehensive Review of Spino-cerebellar Ataxia Type 2 in Cuba. Cerebellum, 10, 184-198. https://doi.org/10.1007/s12311-011-0265-2
- 2. Durr, A. (2010) Autosomal Dominant Cerebellar Ataxias: Polyglutamine Expansions and Beyond. The Lancet Neurology, 9, 885-894. https://doi.org/10.1016/S1474-4422(10)70183-6
- 3. Duenas, A.M., Goold, R. and Giunti, P. (2006) Molecular pathogenesis of Spinocerebellar Ataxias. Brain, 129, 1357-1370. https://doi.org/10.1093/brain/awl081
- 4. Paulson, H.L. (2009) The Spinocerebellar Ataxias. Journal of Neu-ro-Ophthalmology, 29, 227-237. https://doi.org/10.1097/WNO0b013e3181b416de
- 5. Schols, L., Bauer, P., Schmidt, T., et al. (2004) Autosomal Dominant Cerebellar Ataxias: Clinical Features, Genetics, and Pathogenesis. The Lancet Neurology, 3, 291-304. https://doi.org/10.1016/S1474-4422(04)00737-9
- 6. Wadia, N.H. and Swami, R.K. (1971) A New Form of He-redo-Familial Spinocerebellar Degeneration with Slow Eye Movements (Nine Families). Brain, 94, 359-374. https://doi.org/10.1093/brain/94.2.359
- 7. Alonso, E., Martinez-Ruano, L., De Biase, I., et al. (2007) Distinct Distribution of Autosomal Dominant Spinocerebellar Ataxia in the Mexican Population. Movement Disorders, 22, 1050-1053. https://doi.org/10.1002/mds.21470
- 8. Sulek-Piatkowska, A., Zdzienicka, E., Raczynska-Rakowicz, M., et al. (2010) The Occurrence of Spinocerebellar Ataxias Caused by Dynamic Mutations in Polish Patients. Neurolo-gia i Neurochirurgia Polska, 44, 238-245. https://doi.org/10.1016/S0028-3843(14)60037-2
- 9. Gispert, S., Twells, R., Orozco, G., et al. (1993) Chromo-somal Assignment of the Second Locus for Autosomal Dominant Cerebellar Ataxia (SCA2) to Chromosome 12q23-24.1. Nature Genetics, 4, 295-299. https://doi.org/10.1038/ng0793-295
- 10. Imbert, G., Saudou, F., Yvert, G., et al. (1996) Cloning of the Gene for Spinocerebellar Ataxia 2 Reveals a Locus with High Sensitivity to Expanded CAG/Glutamine Repeats. Nature Genetics, 14, 285-291. https://doi.org/10.1038/ng1196-285
- 11. Pulst, S.M., Nechiporuk, A., Nechiporuk, T., et al. (1996) Moderate Ex-pansion of a Normally Biallelic Trinucleotide Repeat in Spinocerebellar Ataxia Type 2. Nature Genetics, 14, 269-276. https://doi.org/10.1038/ng1196-269
- 12. Sanpei, K., Takano, H., Igarashi, S., et al. (1996) Identification of the Spinocerebellar Ataxia Type 2 Gene Using a Direct Identification of Repeat Expansion and Cloning Technique, DIRECT. Nature Genetics, 14, 277-284. https://doi.org/10.1038/ng1196-277
- 13. Velazquez, P.L., Cruz, G.S., Santos, F.N., et al. (2009) Molecular Epide-miology of Spinocerebellar Ataxias in Cuba: Insights into SCA2 Founder Effect in Holguin. Neuroscience Letters, 454, 157-160. https://doi.org/10.1016/j.neulet.2009.03.015
- 14. Cancel, G., Durr, A., Didierjean, O., et al. (1997) Molecular and Clinical Correlations in Spinocerebellar Ataxia 2: A Study of 32 Families. Human Molecular Genetics, 6, 709-715. https://doi.org/10.1093/hmg/6.5.709
- 15. Geschwind, D.H., Perlman, S., Figueroa, C.P., et al. (1997) The Preva-lence and Wide Clinical Spectrum of the Spinocerebellar Ataxia Type 2 Trinucleotide Repeat in Patients with Autosomal Dominant Cerebellar Ataxia. The American Journal of Human Genetics, 60, 842-850.
- 16. Di Fabio, R., Santorelli, F., Bertini, E., et al. (2012) Infantile Childhood Onset of Spinocerebellar Ataxia Type 2. Cerebellum, 11, 526-530. https://doi.org/10.1007/s12311-011-0315-9
- 17. Amarante, T., Takeda, S., Teive, H., et al. (2017) Impact of Dis-ease Duration on Functional Status of Patients with Spinocerebellar Ataxia Type 2. Arquivos de Neuro-Psiquiatria, 75, 773-777. https://doi.org/10.1590/0004-282x20170146
- 18. Estrada, R., Galarraga, J., Orozco, G., et al. (1999) Spino-cerebellar Ataxia 2 (SCA2): Morphometric Analyses in 11 Autopsies. Acta Neuropathologica, 97, 306-310. https://doi.org/10.1007/s004010050989
- 19. Martin, J.J., Krols, L., Ceuterick, C., et al. (1994) On an Autosomal Dominant Form of Retinal-Cerebellar Degeneration: An Autopsy Study of Five Patients in One Family. Acta Neuropa-thologica, 88, 277-286. https://doi.org/10.1007/BF00310370
- 20. Marzi, C., Ciulli, S., Giannelli, M., et al. (2018) Structural Complexity of the Cerebellum and Cerebral Cortex Is Reduced in Spinocerebellar Ataxia Type 2. Journal of Neuroimaging, 28, 688-693. https://doi.org/10.1111/jon.12534
- 21. Mascalchi, M. and Vella, A. (2018) Neuroimaging Applications in Chronic Ataxias. International Review of Neurobiology, 143, 109-162. https://doi.org/10.1016/bs.irn.2018.09.011
- 22. Reetz, K., Rodriguez-Labrada, R., Dogan, I., et al. (2018) Brain Atrophy Measures in Preclinical and Manifest Spinocerebellar Ataxia Type 2. Annals of Clinical and Translational Neu-rology, 5, 128-137. https://doi.org/10.1002/acn3.504
- 23. Yoshii, F., Tomiyasu, H., Watanabe, R., et al. (2017) MRI Signal Abnormal-ities of the Inferior Olivary Nuclei in Spinocerebellar Ataxia Type 2. Case Reports in Neurology, 9, 267-271. https://doi.org/10.1159/000481303
- 24. Orozco, G., Estrada, R., Perry, T.L., et al. (1989) Dominantly Inherited Olivopontocerebellar Atrophy from Eastern Cuba. Clinical, Neuropathological, and Biochemical Findings. Journal of the Neurological Sciences, 93, 37-50. https://doi.org/10.1016/0022-510X(89)90159-7
- 25. Egorova, P.A. and Bezprozvanny, I.B. (2019) Molecular Mechanisms and Therapeutics for Spinocerebellar Ataxia Type 2. Neurotherapeutics, 16, 1050-1073. https://doi.org/10.1007/s13311-019-00777-6
- 26. Kim, Y.E., Jeon, B., Farrer, M.J., et al. (2017) SCA2 Family Presenting as Typical Parkinson’s Disease: 34 Year Follow Up. Parkinsonism & Related Disorders, 40, 69-72. https://doi.org/10.1016/j.parkreldis.2017.04.003
- 27. Sasaki, H., Fukazawa, T., Wakisaka, A., et al. (1996) Central Phenotype and Related Varieties of Spinocerebellar Ataxia 2 (SCA2): A Clinical and Genetic Study with a Pedigree in the Japanese. Journal of the Neurological Sciences, 144, 176-181. https://doi.org/10.1016/S0022-510X(96)00225-0
- 28. Gwinn-Hardy, K., Chen, J.Y., Liu, H.C., et al. (2000) Spinocerebellar Ataxia Type 2 with Parkinsonism in Ethnic Chinese. Neurology, 55, 800-805. https://doi.org/10.1212/WNL.55.6.800
- 29. Lu, C., Chang, H., Kuo, P., et al. (2004) The Parkinsonian Phenotype of Spinocerebellar Ataxia Type 3 in a Taiwanese Family. Parkinsonism & Related Disorders, 10, 369-373. https://doi.org/10.1016/j.parkreldis.2004.03.009
- 30. Shan, D.E., Liu, R.S., Sun, C.M., et al. (2004) Presence of Spinocerebellar Ataxia Type 2 Gene Mutation in a Patient with Apparently Sporadic Parkinson’s Disease: Clinical Impli-cations. Movement Disorders, 19, 1357-1360. https://doi.org/10.1002/mds.20212
- 31. Auburger, G.W. (2012) Spinocerebellar Ataxia Type 2. In: Handbook of Clinical Neurology, Vol. 103, Elsevier, Amsterdam, 423-436. https://doi.org/10.1016/B978-0-444-51892-7.00026-7
- 32. Perez-Avila, I., Fernandez-Vieitez, J.A., Martinez-Gongora, E., et al. (2004) Effects of a Physical Training Program on Quantitative Neurological Indices in Mild Stage Type 2 Spinocerebelar Ataxia Patients. Revue Neurologique, 39, 907-910. https://doi.org/10.33588/rn.3910.2004331
- 33. Scoles, D.R. and Pulst, S.M. (2018) Spinocerebellar Ataxia Type 2. In: Nóbrega, C. and de Almeida, L.P., Eds., Polyglutamine Disorders, Advances in Experimental Medicine and Biology, Vol. 1049, Springer, Berlin, 175-195. https://doi.org/10.1007/978-3-319-71779-1_8
NOTES
*通讯作者。