Applied Physics
Vol.08 No.05(2018), Article ID:25028,6
pages
10.12677/APP.2018.85026
Theoretical Study on the Reaction of C2H2+O2→CH2+CO2
Cunhai Liu, Fenfen Liu, Ye Liu, Bing Guo
School of Basic Sciences for Aviation, Naval Aviation University, Yantai Shandong

Received: May 1st, 2018; accepted: May 17th, 2018; published: May 24th, 2018

ABSTRACT
In this paper, the reaction of small molecule C2H2+O2→CH2+CO2 is researched at the level of B3LYP/6-31G(d,p), using the method of quantum calculation, and the structures of reactant, transition states, intermediates and products, as well as the vibrational modes are gained. The characteristics of each structure in the reaction process are compared and analyzed. The reaction process is confirmed by IRC analysis, and the reaction barrier is obtained. At last, the charge distribution characters of each structure is analyzed.
Keywords:Acetylene, Transition State, Imaginary Frequency, Reaction Barrier
C2H2+O2→CH2+CO2反应的理论研究
刘存海,刘芬芬,柳叶,郭冰
海军航空大学航空基础学院,山东 烟台
收稿日期:2018年5月1日;录用日期:2018年5月17日;发布日期:2018年5月24日

摘 要
本文利用量化计算的方法在B3LYP/6-31G(d,p)水平上对C2H2+O2→CH2+CO2小分子反应过程进行了研究,计算得到了反应物、过渡态、中间体和产物的分子构型,以及过渡态的振动模式,并对反应过程各构型的变化特点进行了对比分析。通过IRC解析的方法对反应过程进行了确认,获得了反应势垒。最后对各构型中原子上电荷分布特点进行了分析。
关键词 :乙炔,过渡态,虚频,势垒

Copyright © 2018 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 引言
1836年,英国皇家学院化学教授戴维•爱德蒙德在加热木炭和碳酸钾以制取金属钾的过程中,将残渣投进水中,产生一种气体,发生爆炸,分析确定这一气体的化学组成是C、H,这种气体就是乙炔。乙炔分子式为C2H2,俗称风煤和电石气,是炔烃化合物系列中体积最小的一员。乙炔在室温下是一种无色、极易燃的气体,乙炔是一种有毒气体,化学性质非常活泼、极易燃、具有爆炸等风险 [1] 。工业上是在装满石棉等多孔物质的钢桶或钢罐中,使多孔物质吸收丙酮后将乙炔压入,以便贮存或运输。纯乙炔是无臭的,但工业用乙炔由于含有硫化氢、磷化氢等杂质,而有一股大蒜的气味。当乙炔在氧气中燃烧时可产生高温和强光,常被用于金属焊接或切割。Laskin和Wang [2] 曾提出乙炔在氧气中燃烧时,C2H2分子可能经过异构化过程转变成•C2H2之后与O2发生反应,并根据实验检测出的产物推测出了诸多反应过程。
过渡态理论是1935年由艾林(Eyring)和波兰尼(Polany)等人提出地,它以统计热力学和量子力学为基础,理论认为由反应物分子转变为生成物分子的过程中间,一定要经过一能级较高的活化络合物,即过渡态,过渡态极不稳定,可以分解为原始的反应物,也可以分解生成产物 [3] 。一直以来,该理论已被广泛应用于对化学过程的研究 [4] - [9] 。本文运用Gaussian 09计算软件,对C2H2+O2→CH2+CO2的反应过程进行了较为系统的计算研究,得出了反应过程中各驻点的几何结构及能量,并利用内禀反应坐标IRC (intrinsic reaction coordinate)的方法对反应物、活性中间体、过渡态和产物之间的联系进行了确认,并依据计算结构对反应过程中分子的结构变化、能量变化和密立根电荷分布进行了分析,以期能为更好的了解乙炔的燃烧过程提供理论支持。
2. 计算方法
理化计算采用的是Gaussian 09计算软件,分子构型软件采用的是GaussView。首先,利用GaussView构型软件构建了反应物、产物、过渡态以及反应中间体的初始构型,然后利用Gaussian09计算软件,在B3LYP/6-31G(d,p)水平上对各初始构型进行了结构优化和频率分析,获得了反应物、产物、过渡态以及反应中间体的优化构型,经频率验证确定优化获得的反应物、产物和反应中间体没有虚频,即构型为稳定构型,优化获得的过渡态构型只有一个虚频,即该构型为C2H2+O2→CH2+CO2小分子反应的中间过渡态。为了确定该分子反应过程中的反应物、产物、过渡态以及反应中间体之间的对应关系,还在B3LYP/6-31G(d,p)水平上进行了内禀反应坐标计算。然后对经IRC内禀反应坐标计算所获得的各中间体的能量进行精确计算,并以反应物作为能量零点得到了各中间体的相对能量,从而绘制出了反应的势能面图。此外,还依据计算结果对反应过程中各结构的密立根电荷分布规律进行了研究。文中能量计算是在MP2/6-311G(d,p)水平上进行地,零点能和频率计算分别采用0.9613和0.8929的修正因子进行校正。
3. 结果与讨论
3.1. 分子的结构特点
图1中列出了优化得到的反应物、过渡态TS1、中间体INT1、过渡态TS2、中间体INT2和产物的几何构型。经频率分析,优化得到的过渡态TS1和过渡态TS2的均有唯一的虚振动频率,从而验证了优化所得过渡态的真实性。首先,反应物C2H2和O2形成反应物络合物,然后O2分子中的5号O原子进攻4号C原子形成过渡态TS1,由于过渡态的不稳定性,5号O原子继续进攻4号C原子,进而成键形成中间体INT1。中间体中的5号O原子向远离4号C原子的方向迁移,6号O原子向4号C原子的方向迁移,从而形成过渡态TS2。同样,TS2具有极不稳定的特性,5号O原子和6号O原子继续向4号C原子迁移,形成INT2,此后INT2中的两个O原子继续向4号C原子两侧移动,并且其中的1C-4C化学键断裂,进而形成产物CH2和CO2。
在反应过程中经历了多次的架键和能量的改变,分子结构也发生的极大变化。图2为反应过程中过渡态TS1、中间体INT1、过渡态TS2和中间体INT2各化学键的变化曲线。不难发现,在由TS1形成INT1过程中2H-1C和3H-1C两个化学键的键长几无变化,而对于4C-1C化学键键长,先减小后有所增加。5O-4C化学键来说,两过渡态中的键长较反应中间体的键长大。而对6O-5O而言,过渡态TS2中的键长最大,而INT1、INT2和TS1中的键长变化不大。表1中列出了中间体和过渡态的键角和二面角参数。比较发现,与键长变化类似,在反应过程中∠3H1C2H和∠4C1C3H未出现较大变化,而∠5O4C1C在TS1中的最小,其余结构中变化不大。从二面角参数来看,反应过程中各原子均在一个平面内。

Figure 1. The structure of each molecule in the reaction process
图1. 反应过程中各分子结构

Figure 2. The change curve of bond length
图2. 键长变化曲线

Table 1. The bond angle and dihedral angle of the intermediate the transition state
表1. 间体和过渡态的键角和二面角
图3为反应过渡态TS1和TS2的振动模式,其中过渡态TS1的振动模式与TS1结构中5O=6O的整体转动相对应,振动频率−1256 cm−1。过渡态TS2的振动模式与TS2结构中两个O原子的面内弯曲振动形成地,其振动频率为−653 cm−1。
3.2. 能量变化分析
采用IRC解析的方法对C2H2+O2→CH2+CO2反应进行了分析,进而确定了优化所得各结构的合理性,并通过对IRC解析所获得的各结构进行了能量计算,通过数据分析获得了该反应的势能曲线,如图4所示。图中横坐标代表反应路径,即反应历程;纵坐标标注的数值是以反应物能量为零参考点所获得的相对能量值。分析发现,首先反应物须跨过8.45 kcal/mol的势垒形成过渡态TS1,然后经1C、4C和5O之间的键重排释放29.98 kcal/mol形成INT1,然后INT1跨过90.82 kcal/mol的势垒,形成TS2,TS2释放239.09 kcal/mol能量后形成中间体INT2。最后INT2经1C、4C和6O之间的价键重排形成产物CH2和CO2。
3.3. 电荷分布
为了更进一步的对C2H2+O2→CH2+CO2反应的过程进行分析,获得了过渡态TS1、中间体INT1、过渡态TS2、中间体INT2中各原子上的密立根电荷分布情况,如图5所示。分析发现,在整个反应过程中

Figure 3. The vibrational modes of transition state
图3. 过渡态振动模式
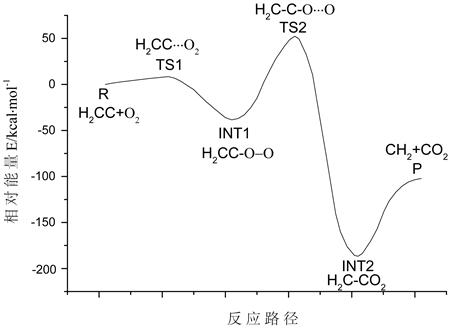
Figure 4. The curve of reaction potential energy
图4. 反应势能曲线

Figure 5. The distribution of Mulliken charge
图5. 密立根电荷分布
1C、2H和3H上的密立根电荷分布变化不大,而对于4C上的电荷分布来看,反应中INT1在形成TS2 过程中密立根电荷没有变化,但在形成TS1的过程中,负电荷逐渐增加,而在由TS2形成INT2的过程中,正电荷出现了明显增加,这主要是由于5O和6O与4C成键的影响。而对5O上的电荷分布来说,随着反应的进行负电荷逐渐增加。此外,在反应过程中6O上的电荷分布TS1上的正电荷最多,然后在形成INT1过程中逐渐减小,然后电荷量又由所增大形成TS2,最后电荷量又减小形成INT2。
4. 结论
对C2H2+O2→CH2+CO2反应的过程进行了分析,对反应过程中各过渡态和中间体,以及反应物和产物的结构变化进行了详细研究。利用IRC解析的方法,对优化所得的各结构的合理性进行了确认,并通过能量分析的方法获得了该反应的势能变化曲线,确定该反应须先后跨过8.45 kcal/mol和90.82 kcal/mol的两个势垒。此外,还对C2H2+O2→CH2+CO2反应的过程中各原子上的密立根电荷分布情况进行了研究,了解了反应过程中各原子上所带电荷的变化过程。
文章引用
刘存海,刘芬芬,柳 叶,郭 冰. C2H2+O2→CH2+CO2反应的理论研究
Theoretical Study on the Reaction of C2H2+O2→CH2+CO2[J]. 应用物理, 2018, 08(05): 216-221. https://doi.org/10.12677/APP.2018.85026
参考文献
- 1. 马欲飞, 佟瑶, 张立功. 基于QEPAS技术的乙炔微量气体高灵敏度检测研究[J]. 光谱学与光谱分析, 2017, 37(9): 2869-2872.
- 2. Gallo, M.M., Hamilton, T.P. and Schaefer, H.F. (1990) Vinylidene: The Final Chapter. Journal of the American Chemical Society, 112, 874-8719. https://doi.org/10.1021/ja00180a011
- 3. 沈玉龙, 舒世立, 刘立华. 现代化学动力学的奠基人——迈克尔•波兰尼[J]. 化学通报, 2016, 79(3): 283-287.
- 4. Song, P., Hu, R. and Zhao, J.F. (2014) Transition State Theory Study on Methyl Butanoate Decomposition Reaction. Journal of Liaoning University (Natural Sciences Edition), 41, 216-221.
- 5. Wu, S.L., Shen, D.K. and Gao, S.Y. (2013) Thermodynamic Analysis and Transition State Study for Pyrolysis of Levoglucosan and Glyceraldehydes through Quantum Simulation. Journal of Southeast University (English Edition), 29, 282-288.
- 6. Wang, Z.J., Chen, M. and Guo, Z.Y. (2002) Analysis of Transition State Theory for Condensation. Chinese Science Bulletin, 47, 952-954.
- 7. Sun, Y.M., Wu, J.S. and Liu, C.B. (2007) A Comparison of Transition State of Phenol in H-Atom Ab-straction by Methyl and Methylperoxyl Radicals. Chinese Science Bulletin, 52, 1182-1186. https://doi.org/10.1007/s11434-007-0172-6
- 8. 王成云, 顾慰中. 配合物氧化还原反应两种机理的过渡态[J]. 曲阜师范大学学报(自然科学版), 2001, 27(1): 59-61.
- 9. 赵衍辉, 刘杰, 张新. 孤立条件下α-丙氨酸分子碳骨架异构的过渡态理论研究[J]. 白城师范学院学报, 2014, 28(3): 6-8.