Hans Journal of Biomedicine
Vol.
09
No.
01
(
2019
), Article ID:
28346
,
6
pages
10.12677/HJBM.2019.91003
Protein Secondary Structure Prediction Based on Wavelet Feature Extraction and Support Vector Machine
Jian Wang, Jinyong Cheng*
College of Information, Qilu University of Technology (Shandong Academy of Sciences), Jinan Shandong

Received: Dec. 16th, 2018; accepted: Dec. 31st, 2018; published: Jan. 7th, 2019

ABSTRACT
The structure of proteins is very important for understanding the biological function of proteins. The prediction of protein structure can predict and understand the function of biological functions of unknown proteins; however, the prediction of protein secondary structure plays a decisive role in the prediction of protein structure. In the study of protein secondary structure prediction, a single residue of a protein is encoded by position-specific-score-matrix (PSSM). After a data window is taken, a protein residue can be represented as a 2-dimensional pseudo-image plane, thus could further use the wavelet method to extract multi-resolution based features both on high frequency and low frequency from original pseudo-image, these extracted wavelet-based features with the PSSM matrix together can be taken as sample information carried by a protein residue, and the training model used is support vector machine.
Keywords:Protein Secondary Structure Prediction, PSSM, Pseudo-Image, Wavelet Transform, Support Vector Machine
基于小波特征提取和支持向量机的蛋白质二级结构预测
王剑,成金勇*
齐鲁工业大学(山东省科学院)信息学院,山东 济南
收稿日期:2018年12月16日;录用日期:2018年12月31日;发布日期:2019年1月7日

摘 要
蛋白质的结构对理解蛋白质的生物学功能意义重大,蛋白质结构的预测就能预测和理解未知蛋白质生物学功能的作用,并且蛋白质二级结构的预测是对蛋白质结构的预测起决定性作用的,在蛋白质二级结构预测的研究中,将蛋白质单个残基用位置特异性打分矩阵(PSSM)进行编码,取窗口后可以将一个蛋白质残基表示成一个2维的伪图像平面,在原位置特异性打分矩阵数据平面的基础上,用小波变换提取到伪图像平面不同分辨率水平上的低频特征和高频特征与原PSSM平面数据当作一个蛋白质残基携带的样本信息,并用支持向量机对预测进行训练模型。
关键词 :蛋白质二级结构预测,位置特异性打分矩阵,伪图像,小波变换,支持向量机

Copyright © 2019 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 引言
蛋白质三维结构和其功能的研究是许多相关领域例如药物和酶的设计和制造的重要研究组成部分 [1] [2] [3] 。蛋白质的三维结构在很大程度上取决于蛋白质序列的氨基酸残基排列 [4] ,这是因为蛋白质氨基酸残基序列包含了蛋白质本身的全部信息。因为无法直接从蛋白质的氨基酸残基序列预测出蛋白质的三维结构 [5] ,所以理解和研究复杂的蛋白质氨基酸残基序列元素的相互关联一直是生物信息学中的巨大挑战 [6] [7] ,也使蛋白质二级结构预测成为蛋白质结构预测的重要环节 [8] [9] [10] [11] 。
蛋白质二级结构是一种局部多个蛋白质氨基酸表现出相对稳定的结构特性,Pauling [12] 在60年前提出有两种常见的蛋白质二级结构类型分别是alpha螺旋,用H表示,与beta折叠,用E表示,还有一种不规则区域结构被称为不规则卷曲用C表示。Sander [13] 开发了一种被称作DSSP算法将蛋白质二级结构总结分成了八种状态,并且将其中三种指定为螺旋(H)类型,其中两种指定为折叠(E)类型,其他剩余部分指定为卷曲(C)类型。
在蛋白质二级结构预测的研究中,通常多数研究者会将蛋白质残基编码成位置特异性打分矩阵(PSSM),将PSSM应用于蛋白质二级结构预测开始于Jones [14] 。PSSM是替代打分矩阵发展而来的,是序列联配促使产生了替代打分矩阵,例如BLOSUM多重进化矩阵 [15] 。PSSM序列联配打分考虑了序列比对中的位置信息,在序列联配时对每个比对位点作出了独立的打分来表现这些位点的偏好。
本文在PSSM蛋白质残基编码的基础上,将输入样本增加了小波变换后的获得的伪图像特征和细节特征 [16] 对支持向量机蛋白质二级结构预测做了研究。
2. 蛋白质残基的编码方法
本文中将单个蛋白质残基结构序列用PSSM编码,此外还添加了正交编码来记录蛋白质氨基酸的类型,其中一个蛋白质残基包含20维PSSM信息和20维残基类型信息,氨基酸序列取窗口大小为15表示中间的残基,这样以来每个蛋白质残基被表示成了[40 * 15]的数据平面。
3. 小波分析提取PSSM的特征
3.1. 小波变换的简要阐述
小波分析方法是一种利用小波函数 [17] 作为基函数和尺度函数 [18] 的对信号波进行信号成分分析的方法,假设有信号
(1)
( 是尺度系数, 是小波系数),公式(1)右边第一项表示信号在尺度空间得低频信号,第二项代表小波分解到的高频特征分别用v和w表示,一个 信号可以被无限二分解为 和 , , ,利用小波变换可以得到原信号的一个尺度空间的平滑近似和小波空间的小波系数分别对应公式(1)的右边第一项和第二项。
3.2. 不同分别率下图像的特征
高分辨率的图像会在低分辨率下形成一个原图像的近似 [16] ,低分辨率下的图像与原图像之间的差别被看作是原图像的细节特征,这种细节特征和图像近似可以利用二维小波变换来实现转换提取,一个图像在小波变换第n层次的分解获得的低频特征就是这个图像在第n低分辨率下的图像近似,当然高频特征就是细节特征。
由此本文尝试对残基PSSM数据平面进行小波变换,将蛋白质残基PSSM视作伪图像平面,在第二个分辨率水平上提取到图像近似和细节特征作为原PSSM样本的辅助特征,小波变换的小波函数设置为“haar”小波,辅助特征提取示意图如图1所示:
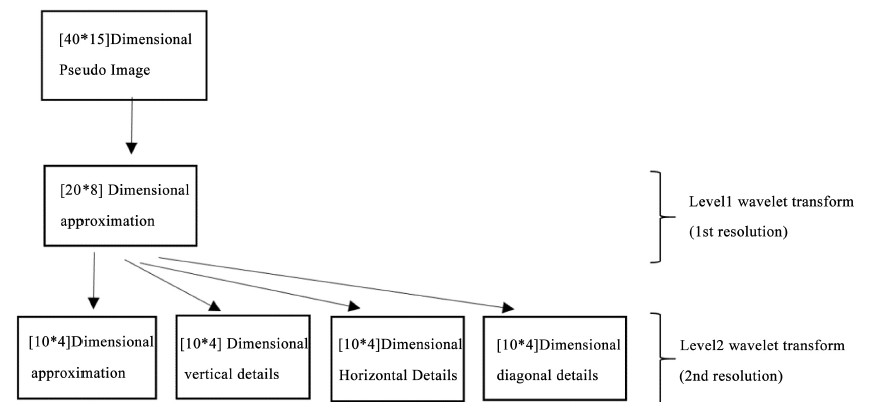
Figure 1. Extract multiresolution features use wavelet transform from PSSM
图1. 从PSSM中提取不同分辨率水平下的伪图像特征
4. 支持向量机的引入
4.1. 支持向量机的简单阐述
对于样本空间 ,对应标签 ,找到一个分类超平面使X满足 ,使得正负样本在被分类平面分开,并且求解 ,找到最
大间隔分类面,其中 是松弛变量,它的出现可以使支持向量可以分布在分类边缘的周围,使得最大间隔分类面得到了充分实现,w权值记录了训练样本中支持变量的影响权重,C参数控制了离群样本对超平面选择的影响, 是一个核函数将原样本空间映射到高维数据空间避免了线性不可分的情况 [19] 。
4.2. 从支持向量得到的启发
本文考虑到样本的特征提取会影响到样本空间中支持向量的有效选取 [20] ,所以在原来样本特征的基础上,添加了小波变换产生的高维特征和低维特征,使得支持向量机在运行SMO算法 [21] 时能够更加有效的填充w权值使得支持向量可以更加合理的分布在超平面附近达到更好的分类效果。
4.3. 支持向量机的输入输出和参数
支持向量机的输入一共包含两部分,第一是[40 * 15]维度的PSSM+正交编码,第二是4个[10 * 4]维度的数据平面,两部分相加,支持向量机的输入是一个760维的包含PSSM+正交编码和小波变换的向量。用Matlab2017b的支持向量机工具做了实验,支持向量机的核函数有“gaussian”,“linear”,和“polynomial”,本文中设置为多项式函数(polynomial),支持向量机的类别划分方法有两种,分别是“one vs all”和“one vs one”,本文中设置为“one vs one”,支持向量机的预测标签设置为DSSP算法定义的三类二级结构标签分别为卷曲(C),螺旋(H)和折叠(E)。
5. 预测结果的评估方法
对于蛋白质二级结构预测结果的评估本文采用的是Q3评估方法,在Q3的计算过程中,文中采用了三折交叉验证(3-fold cross validation),关于Q3评估方法的描述如下:
,其中 分别是H类E类C类残基预测正确的残基数量。
, ,
6. 试验结果与讨论
6.1.试验结果展示
本文对PSSM样本矩阵与PSSM样本矩阵 + 小波变换辅助特征的SVM试验结果都做了记录,如表1:

Table 1. Q3 accuracy comparison between two support-vector-machine-based methods
表1. 两种支持向量机预测方法Q3正确率的比较
通过观察表1结果可以发现,小波变换取得的辅助特征增加了螺旋(H)和折叠(E)的预测正确率,同时对卷曲的预测结果有一定程度上的缩减,不过卷曲正确率的缩减原没有超过螺旋(H)和折叠(E)正确率提升对Q3正确率的影响,所以在添加小波变换后的特征之后相比于只有PSSM矩阵,正确率提高了1.9%。
6.2. 分析讨论
本文将PSSM矩阵当作伪图像之后,可以方便利用二维小波变换进行低分辨率水平的特征提取,文中在第二个分辨率水平上对PSSM伪图像进行了近似特征(分辨率水平上的低频特征)和三种细节特征(三种高频特征,分别是横向,纵向,斜向)的提取,这样做的目的是我们期望将PSSM所包含的蛋白质残基特征利用小波变换在另一个数据空间突显出来,并保证特征效果不低于原来的PSSM矩阵。所以我们将小波变换提取到的特征与PSSM组合组成了新的样本特征矩阵。
基于对支持向量机的研究,其是根据数据样本特征空间找到合适的支持向量,在数据样本特征足够的情况下有利于支持向量机的功能实现,从实验结果看来,三种类别的蛋白质残基预测结果相对于小波变换之前除了 之外有稍微下降,其余两类有显著的提升,这说明小波变换可以提取出关于PSSM的另一种数据特征作为数据样本特征输入支持向量机。
7. 总结
本文为了充分利用支持向量机的分类特性,为了使蛋白质残基能携带能充分表示一个蛋白质残基的特征,本文中引入了小波变换将PSSM数据平面的高频特征和低频特征作为PSSM数据平面的辅助特征,通过观察实验结果,Q3正确率得到了明显提升。
基金项目
国家自然科学基金(61375013);山东省自然科学基金(ZR2013FM020)。
文章引用
王 剑,成金勇. 基于小波特征提取和支持向量机的蛋白质二级结构预测
Protein Secondary Structure Prediction Based on Wavelet Feature Extraction and Support Vector Machine[J]. 生物医学, 2019, 09(01): 17-22. https://doi.org/10.12677/HJBM.2019.91003
参考文献
- 1. Petsko, G.A. and Ringe, D. (2002) Protein Structure and Function. Lorne Protein Workshop.
- 2. Whittle, P.J. and Blundell, T.L. (1994) Protein Structure-Based Drug Design. Annual Review of Biophysics and Biomolecular Structure, 23, 349-375. https://doi.org/10.1146/annurev.bb.23.060194.002025
- 3. Schaffhausen, J. (2012) Advances in Structure-Based Drug Design. Trends in Pharmacological Sciences, 33, 223. https://doi.org/10.1016/j.tips.2012.03.011
- 4. Baker, D. and Sali, A. (2001) Protein Structure Prediction and Structural Genomics. Science, 294, 93-96. https://doi.org/10.1126/science.1065659
- 5. Dill, K.A. and Maccallum, J.L. (2012) The Protein-Folding Problem, 50 Years on. Science, 338, 1042-1046. https://doi.org/10.1126/science.1219021
- 6. Whisstock, J.C. and Lesk, A.M. (2003) Prediction of Protein Func-tion from Protein Sequence and Structure. Quarterly Reviews of Biophysics, 36, 307. https://doi.org/10.1017/S0033583503003901
- 7. Lee, D., Redfern, O. and Orengo, C. (2007) Predicting Protein Function from Sequence and Structure. Nature Reviews Molecular Cell Biology, 8, 995-1005. https://doi.org/10.1038/nrm2281
- 8. Radivojac, P., Clark, W.T., Oron, T.R., et al. (2013) A Large-Scale Evalu-ation of Computational Protein Function Prediction. Nature Methods, 10, 221. https://doi.org/10.1038/nmeth.2340
- 9. Lin, K., Simossis, V.A., Taylor, W.R., et al. (2004) A Simple and Fast Secondary Structure Prediction Method Using Hidden Neural Networks. Bioinformatics, 21, 152-159. https://doi.org/10.1093/bioinformatics/bth487
- 10. Yoo, P.D., Zhou, B.B. and Zomaya, A.Y. (2008) Machine Learning Techniques for Protein Secondary Structure Prediction: An Overview and Evaluation. Current Bioinformatics, 3, 74-86. https://doi.org/10.2174/157489308784340676
- 11. Faraggi, E., Zhang, T., Yang, Y., et al. (2012) SPINE X: Improving Protein Secondary Structure Prediction by Multistep Learning Coupled with Prediction of Solvent Accessible Surface Area and Backbone Torsion Angles. Journal of Computational Chemistry, 33, 259-267. https://doi.org/10.1002/jcc.21968
- 12. Pauling, L., Corey, R.B. and Branson, H.R. (1951) The Structure of Proteins: Two Hydrogen-Bonded Helical Configurations of the Polypeptide Chain. Proceedings of the National Academy of Sciences, 37, 205-211. https://doi.org/10.1073/pnas.37.4.205
- 13. Kabsch, W. and Sander, C. (1983) Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers, 22, 2577-2637. https://doi.org/10.1002/bip.360221211
- 14. Jones, D.T. (1999) Protein Secondary Structure Prediction Based on Position-Specific Scoring Matrices. Journal of Molecular Biology, 292, 195-202. https://doi.org/10.1006/jmbi.1999.3091
- 15. Henikoff, S. and Henikoff, J.G. (1992) Amino Acid Substitution Matrices from Protein Blocks. Proceedings of the National Academy of Sciences, 89, 10915-10919. https://doi.org/10.1073/pnas.89.22.10915
- 16. Mallat, S.G. (1989) A Theory for Multiresolution Signal Decom-position: The Wavelet Representation. IEEE Transactions on Pattern Analysis and Machine Intelligence, 11, 674-693. https://doi.org/10.1109/34.192463
- 17. 于开平, 邹经湘, 杨炳渊. 小波函数的性质及其应用研究[J]. 哈尔滨工业大学学报, 2000, 32(2): 36-39.
- 18. 丁宣浩. 由尺度函数构造小波的一个充要条件[J]. 工程数学学报, 2007, 24(2): 273-281.
- 19. 张铃. 基于核函数的SVM机与三层前向神经网络的关系[J]. 计算机学报, 2002, 25(7): 696-700.
- 20. 汪廷华, 田盛丰, 黄厚宽, 等. 样本属性重要度的支持向量机方法[J]. 北京交通大学学报, 2007, 31(5): 87-90.
- 21. 张召, 黄国兴, 鲍钰. 一种改进的SMO算法[J]. 计算机科学, 2003, 30(8): 128-129.
NOTES
*通讯作者。