Hans Journal of Computational Biology
Vol.4 No.02(2014), Article ID:13671,9 pages
DOI:10.12677/HJCB.2014.42004
Construction of Molecular Biology Secondary Database Resources Platform
1School of Computer Science and Technology, Xinjiang Normal University, Urumqi
2College of Computer Science, Yangtze University, Jingzhou
Email: 563988413@qq.com, *757593664@qq.com
Copyright © 2014 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received: May 14th, 2014; revised: May 20th, 2014; accepted: May 29th, 2014
ABSTRACT
In bioinformatics, bio-secondary database can build more in-depth study of specific species. Response to the urgent need to build a platform for molecular biology secondary database resources, molecular biology secondary database was constructed by using biosql; using tomcat 6.0 + Myeclipse + mysql technology and MVC development model in order to achieve a gene as an example of resilience web resource database platform. The platform is built to solve the molecular biology and genetic data resilience secondary database obtain automatically, researchers can receive timely retrieval of data quickly upload and resilience genes. The platform can also be applied to other types of molecular biology to accelerate research on a range of molecular biology, making research more targeted.
Keywords:Bioinformatics, Molecular Biology Secondary Database, BioPerl, MVC Model

分子生物学二次数据库资源平台的构建
杜 晶1,曹兴芹2*
1新疆师范大学计算机科学技术学院,乌鲁木齐
2长江大学计算机科学学院,荆州
Email: 563988413@qq.com, *757593664@qq.com
收稿日期:2014年5月14日;修回日期:2014年5月20日;录用日期:2014年5月29日

摘 要
在生物信息学中,建立生物二次数据库可以针对特定物种进行更深入的研究。针对分子生物学二次数据库资源平台的构建的迫切需要,利用biosql构建了分子生物学二次数据库,运用tomcat 6.0 + Myeclipse + mysql技术,MVC开发模式,实现了以抗逆基因为例的web资源数据库平台。该平台解决了分子生物二次数据库的构建与抗逆基因数据自动获取,能及时接受研究者数据的上传和抗逆基因的快速检索。该平台同样可以应用到其他类别的分子生物,加快了对某一范围内分子生物学的研究,使研究更具针对性。
关键词
生物信息学,分子生物二次数据库,BioPerl,MVC模型

1. 引言
生物信息学就其本身字面意义是所有关于生物的信息研究的学科。但是一般意义上的生物信息学就专门指关于分子水平的各种生物大分子序列、结构和功能上的信息研究。分子生物信息数据库主要包括基因组数据库、核酸和蛋白质序列数据库、蛋白质构数据库等初始数据库,以及由此而构建的二次数据库、复合数据库,即面向生物学家具有各种不同特色或特殊用途的专门数据库[1] 。自人类基因组计划从20世纪90年代开始启动后,对分子生物研究与日俱增,然而相关数据库资源平台却屈指可数。一般而言,生物信息数据库可以分为一级数据库和二级数据库。一级数据库的数据都直接来源于实验获得的原始数据,只经过简单的归类整理和注释[2] 。国际上著名的三大核酸数据库NCBI的Genebank、日本的DDBJ和EBI的EMBL数据库都是建立二次数据库的数据来源。Bioperl作为Perl语言专门用于生物信息的工具与函数模块集,是世界各地的perl开发在生物信息学、基因组学以及其他生命科学领域的智能结晶[3] 。利用Bioperl构建分子数据库是研究人员广泛使用的方法,已经形成了行业标准。
事实上,分子生物学家一般仅仅需要某一类分子生物学数据,仅从一级数据库查找所需要的数据形如大海捞针,因此建立本地二次数据库筛选并存储所需要的数据的实现具有重大意义。二次数据库是在一级数据库、实验数据和理论分析的基础上针对特定目标衍生而来的,是对生物学知识和信息的进一步整理。
如何建立高质量的二次数据资源平台,从最新国内浙江大学白琳于2012年建立了植物抗逆基因资源平台的构建[4] 和国外知名网站研究进展来看,还没有的到很好解决。因此本平台基于以抗逆基因为例,在实现了高性能的抗逆基因的检索算法下立了二次数据库,并利用WEB技术建立适合广大研究者使用的WEBRGSBD。相对于其平台而言,本系统实现了数据的用户上传和自动获取且信息的显示更加充分的能按NCBI的genbank格式要求,方便研究人员分析数据。
2. 分子生物二次数据库构建
2.1. 数据库基本设计思路
早期,大多数生物信息学和计算生物学的应用程序开发都是基于unix/Linux环境中,bioperl作为Perl的生物模块,开发人员开发了大量基于unix/Linux处理分子生物序列数据。目前而言bioperl也将其中的一些程序移植到Windows,便于生物学研究者在没有编程背景下进行生物信息学分析[5] 。但是程序功能完备性不如Linux上。因此本资源平台的二次数据库在linux发行的centos 5.3版本上构建的。在linux平台上安装mysql数据库,bioperl程序与mysql的接口文件DBI和DBD,以及bioperl的相关生物信息功能处理模块,最后导入从BioSQL网站下载biosqldb-mysql.sql文件,构建分子生物分子数据库模型。
2.2. 数据的收集和整理
要建立二次数据库,首先必须准备相关数据源,即获取相应的生物数据。主要途径包括:
1) 利用bioperl脚本程序从远程下载大批量数据。生物分子数据量巨大,特别是核酸序列的数据以千兆计,有组织地搜集和管理这些数据已成为生物信息学研究的主要内容之一[6] 。向福于2004年开发了基于基因序列获取的程序设计[7] ,其获取仅针对基因序列号下载,不能获得精准的指定类别的基因序列。
2) 使用者的研究数据。分子生物学的研究进展缓慢的一个主要原因是资源的封闭性。例如DRASTIC INSIGHTS网站是目前比较认可的抗逆基因胁迫分析平台,但其主要提供分析的结果显示,不能提供用户数据上传,因此信息量较小,更新速度较慢。
3) 基于web网页从一级数据库(如NCBI的Genbank)中手工下载。国际上著名的三大核酸数据库NCBI的Genebank、日本的DDBJ和EBI的EMBL数据库面向全世界提供数据上传,下载,分析功能,但其数据过多,未较好的分类。以此获取分子生物学二次数据库所需数据耗时,耗力,效率不高。
本平台采用前2种途径:1) 在文献中提出某一类基因的关键字基础上,实现下载所需大批量数据的算法,该算法非常方便,且通用性强,只需将这算法中关键词换掉,便可下载所需序列。2) 使用者把自己的研究数据通过web服务传到服务器,经管理员通过后台上传到数据库中。
2.3. 构建分子生物二次数据库
biosql是生物信息学界构建二次数据库比较通用的一个数据模型,其覆盖序列,特征,序列和功能注释,参考分类学和本体(或受控词表),其整合了多个公共数据库资源的数据,包括GenBank,Gene Ontology (GO),taxonomy。[8] 基于biosql构建的数据模型包含28个数据表,但在实际应用中并不是所有的数据表都需要使用,因此是在分子生物二次数据库的构建中对已有的数据模型根据进行重构方便对资源平台的应用开发。本平台使用到的各种数据包及bioperl脚本见表1。
在导入biosqldb-mysql.sql时,linux命令行提示关于错误提示'TYPE = INNODB',由于mysql版本的更新,在MYSQL 5.1之后TYPE不在使用。解决的办法是把biosqldb-mysql.sql文件中的所有“TYPE”换成“engine”。
利用bioperl编写的脚本从ncbi的genbank数据库下载用户所需的数据,导入到本地构建的biosql数据库。根据genbank格式的数据在数据库中的存储位置,抽象出二次分子学数据库所用的数据模型。抽象出的ER图如图1。
GenBank数据库包含基本单位是序列条目,包括核甘酸碱基排列顺序和注释两部分。本文中采用genbank数据库的数据。所抽象出来的数据模型中:bioentry表,bioentry_reference表,reference表,dbxref表通过主外键相互关联描述了序列的名称、长度、日期、序列说明、编号、版本号、相关文献作者、题目、刊物、日期。而location表,bioentry_qualifier_value表,seqfeature表,seqfeature_qualifier_value表,term表,ontology表,biosequence表描述了序列的物种来源、学名、分类学位置、序列特征表、碱基组成。其中序列特征表(Feature table)包含了大量与序列直接相关的注释信息,这些信息为数据库的使用和二次开发提供了基础。gene ontology和taxony可以说是一套用于生物信息数据库的规范体系,在构建数据库时为了使得各个不同的数据库中对于序列的相关注释信息保持一致,便于使用者获得信息的完整性。Gene Ontology中最基本的概念是term。

Table 1. Data packet to build a molecular biology secondary database
表1. 分子生物二次数据库构建数据包
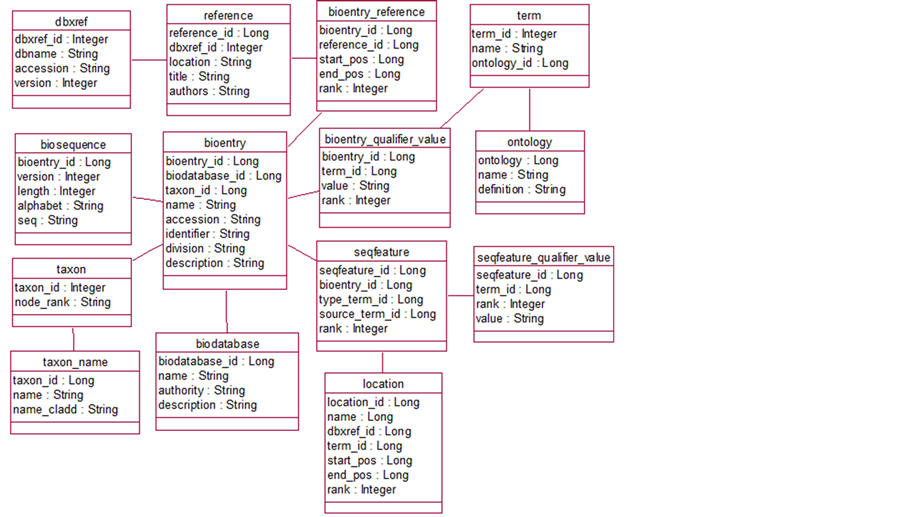
Figure 1. Secondary database E-R diagram
图1. 二次数据库E-R图
在构建了数据库的模型基础上,通过前期已经实现的基于BioPerl实现从NCBI下载基因序列下载到本地,通过bioperl的序列导入模块,通过命令行导入二次数据库。
[root@localhost~]#cd/usr/local/BioPerl-DB-1.006900/scripts/biosql
[root@localhost biosql]#perl./load_seqdatabase.pl-host localhost-dbuser root-dbpass123456-dbname biosql -namespace genbank-format genbank/usr/mysequence1.gb
3. WEB分子生物数据库发布平台
生物信息学研究中的WEB技术包括WEB客户端技术和WEB服务器端技术,两者缺一不可。WE客户端技术实现数据共享,WEB服务器技术很方便的实现对外发布信息和信息交流[9] 。在上述已建立好的生物二次数据库基础上,利用tomcat 6.0 + Myeclipse + mysql相结合,通过MVC (Model-View-Control)三层开发模式,构建了抗逆基因数据库信息平台。MVC是把一个应用的输入、处理、输出流程按照Model-View-Controller的方式进行分离,这样一个应用被分成三个层——模型层、视图层、控制层[10] 。利用这种分层模式能有效合理地开发,实现了“高内聚,低耦合”。此平台的系统结构如图2。
3.1. WEB分子生物数据库发布平台搭建
用于分子生物信息学开发的编程语言越来越多,主流的是Python和PERL,java,php.但如果考虑在web应用开发中的平台可移植性和程序执行速度,java又成为首选。因此java语言开发了很多用于分子生物数据处理的程序,即biojava。目前最新的biojava已经形成一个开源框架致力于提供一个java框架来处理生物数据。在linux平台上开发java WEB程序的集成开发环境是eclipse,但在linux上开发难度大。因此,本资源平台的开发在windows上实现。在linux和windows分别安装并配置java开发环境(jdk)、tomcat环境,把开发好的应用通过tomcat发布。为了在linux访问方便,可把tomcat设置为开机自启动,对linux的根目录下etc下的rc.local文件进行更改[11] ,配置如下:
vi rc.local export JAVA_HOME=/var/ftp/pub/jdk6
/usr/local/tomcat/tomcat6/bin/startup.sh
由于perl在windows上构建分子生物数据库难度较大,但开发又是基于windows.因此把已经构建好的分子生物数据库移植到windows上。开发过程中遇到对于2种平台数据库的完全移植平台之间的差异性,造成移植失败。在因此采用把数据的结构和数据分离。数据库的结构从BioSQL网站下载biosqldb-mysql.sql文件,不用导出,只导入数据到windows上的biosql数据库即可:
导出数据库数据:mysqldump-t biosql-u root-p > biosql.sql
导入数据:source d:\biosql.sql;
这样便可在windows上进行开发,但测试数据有限。数据来源是使用在linux平台下的perl load_seqdatabase.pl脚本导入到数据库中,最后把在windows测试好的应用,发布到linux平台上。
3.2. 平台功能
分子生物学二次数据库的资源平台是一个基于B/S(浏览器/服务器)访问模式,方便广大研究者的。使用该平台主要包括三部分:1) 根据抗逆基因特征,通过perl语言实现所需抗逆基因数据的远程下载到本
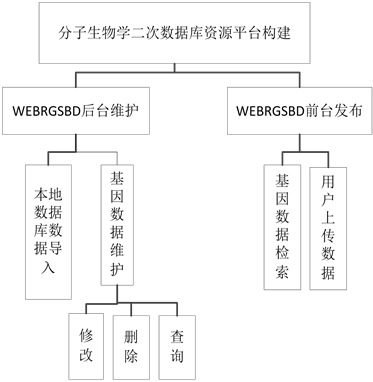
Figure 2. System function module diagram
图2. 系统功能模块图
地。通过此平台提供的文件导入数据库功能,实现数据库存储;2) 互联网上的用户通过文件上传功能上传gebank格式的最新抗逆基因研究成果;3) 用户根据关键字检索相关抗逆基因。搭建的抗逆基因数据库平台的功能如3.1所介绍。下面依次具体介绍这三种功能及主要算法实现:
3.2.1. 数据库数据导入
数据的导入可采用bioperl模块在Linux下通过命令终端完成,但使用起来不方便,易出错。不熟悉编程的人员使用起来更是困难。因此本平台获取导入数据库的文件路径通过script脚本验证文件格式是否正确。把导入数据库的脚本命令利用java中输出流写入到bat文件中,再用java的Runtime和Process类调用另一个现成的可执行程序或系统命令使用Runtime.getRuntime().exec()方法调用系统命令执行bat文件。对于用户而言只需要选择文件即可。
实现数据导入的核心代码如下:
String a=request.getParameter("filename");
Process process;
String s="perl/usr/local/BioPerl-DB-1.006900/scripts/biosql/load_seqdatabase.pl-host localhost-dbuser root -dbpass 123456 -dbname biosql -namespace bioperl -format genbank /usr/local/"+a;
//使用Runtime来执行command,生成Process对象
Runtime runtime = Runtime.getRuntime();
process = runtime.exec(cmd);
3.2.2. 文件上传功能
借助apache的FileUpload包和io包实现文件上传功能,并把解压后的jar包放到所在应用的lib包中。通过后台程序调用实现文件上传,同样限定文件格式为genbank,使用与数据库数据导入相同的脚本验证文件格式。
script脚本验证文件格式:
function isValidate(){
if(document.getElementById("filename").value.replace(/\s/g, "") != "")
{var FileType = "gbk";
//--这里是允许的后缀名,注意要小写
Var FileName= document.getElementById("filename").value; FileName=FileName.substring (FileName. lastIndexOf('.') + 1, FileName.length).toLowerCase();
//--这里把后缀名转为统一转为小写了
if (FileType.indexOf(FileName) = -1){alert("附件格式不正确!");return false;}} return true;}
3.2.3. 信息检索功能
用户根据所需关键字检索抗逆基因信息,检索时忽略字母大小写。结果主要显示基因的序列名称和序列简单说明。
3.2.4. 信息显示
经分析,genbank格式的数据主要显示基因的基本信息,来源,序列信息,特征部分四部分信息。本系统实现的信息检索的结果与从NCBI上面下载的gebank格式数据的文本数据内容一致(图3)。由于其数据在数据库的存储涉及多张表,关联关系较为复杂,因此建立4个实体类:分别是Bioentry(抗逆基因实体类),Refrence (抗逆基因参考文献),Biosequence (抗逆基因序列),seqfeature_qualifier_value (抗逆基因特征),其中特征表部分需要用到左右链接左匹配,在显示是对应的基因的信息的名词和具体值分别就是sql语句中的st.name,st.value。其sql语句的实现为:
select st.name, st.value from (SELECT t.name,s.value, s.seqfeature_id FROM term t left join seqfeature_qualifier_value s on t.term_id=s.term_id) st inner join (SELECT b.bioentry_id, sq.seqfeature_id FROM bioentry b left join seqfeature sq on b.bioentry_id=sq.bioentry_id where b.name='?') sqb on st.seqfeature_id = sqb.seqfeature_id group by st.name, st.value。
3.2.5. VNC远程访问linux
linux发行的centos5.3版本提供了VNC服务,方便其他计算机对linux操作系统的远程访问,实现了程序员通过windows及时远程查看其运行情况,不用直接到linux服务器上去查看。同样可把vnc设为自启动,方便服务器由于重启后,重新连接vnc.
vi/etc/rc.d/rc.local
使用vi编辑器打开配置文件,并进行下列修改
/etc/init.d/vncserver start——新增行。
3.3. 平台实现及测试
此平台主要实现用户安全登录,管理员上传数据,维护数据,普通用户上传研究结果到服务器由管理员核实后上传,信息检索。本测试以上传基因号为1DH3_B测试器导入,检索,显示功能,具体测试
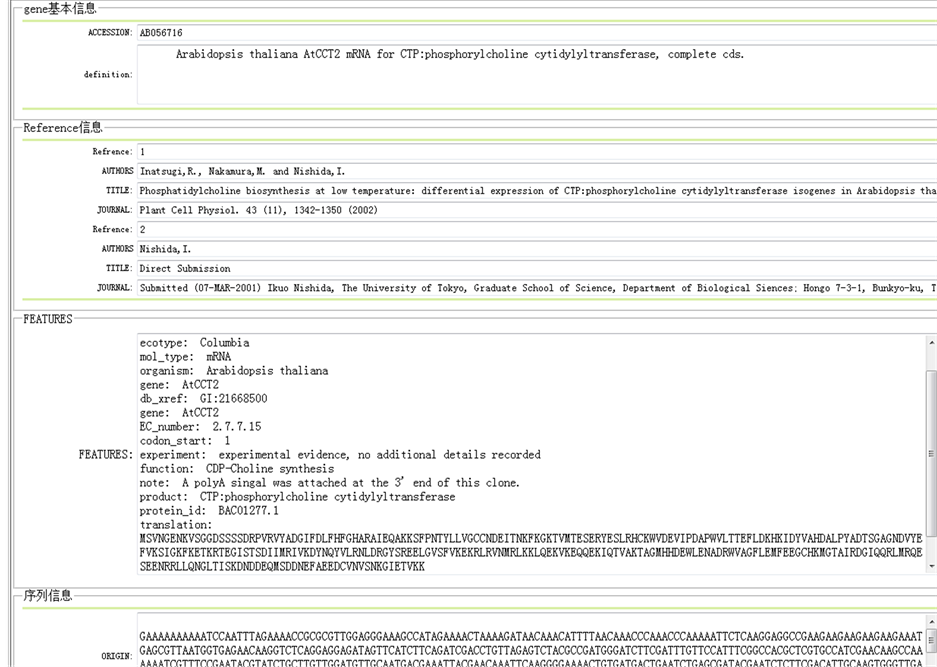
Figure 3. Gene details
图3. 基因信息详细信息
如下:
管理员书输入用户名密码,登陆系统(图4)。后台通过图片动态验证,并匹配数据库中的记录,验证是否为合法用户。并可记住密码,方便用户使用。
管理员通过数据库本地导入功能,选择1dh3_b.gbk格式的上传(图5),上传时字母的大小写忽略。如果不是gbk格式提示信息,如果上传成功则显示后台上传的过程,如图6。
在检索框内输入基因的名字1dh3_b,检索出基因记录(图7),并点击详细按钮,可查看本条记录的详细信息(图8)。
此页面显示了基因的基本信息,来源信息,特征信息及序列信息。来源可能涉及多个参考文献,所以采用循环ArrayList集合输出来源信息。每一部分的信息的名字来源于本体表Gene Ontology (GO)中的term表的name字段,对应的具体值来源于bioentry_qualifier_value表的value字段。
用户上传研究的数据到服务器,同样格式要求是genbank格式,如果结果不正确,则提示信息(图9),格式正确,同意保存在服务器的/usr/local/genbank目录下。
以上即为本系统的测试主要测试部分,后面将实现文件的下载和blast序列比对,方便研究者进行研究。

Figure 4. User login
图4. 用户登录
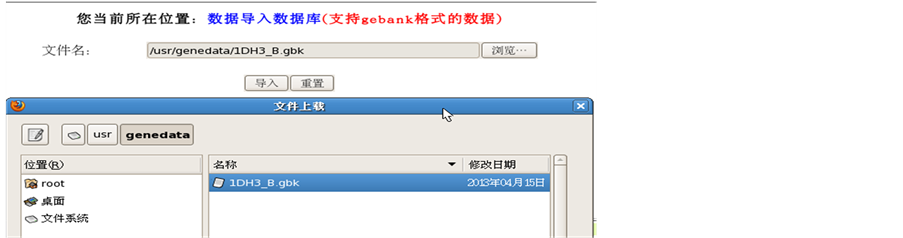
Figure 5. Choose data to upload
图5. 选择上传数据
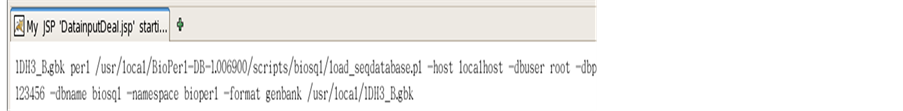
Figure 6. Data into the database
图6. 数据导入数据库

Figure 7. Gene information retrieval results
图7. 基因信息检索结果
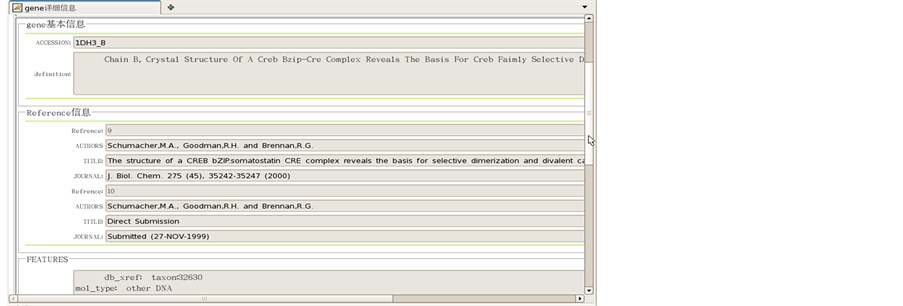
Figure 8. Retrieve details
图8. 检索详情
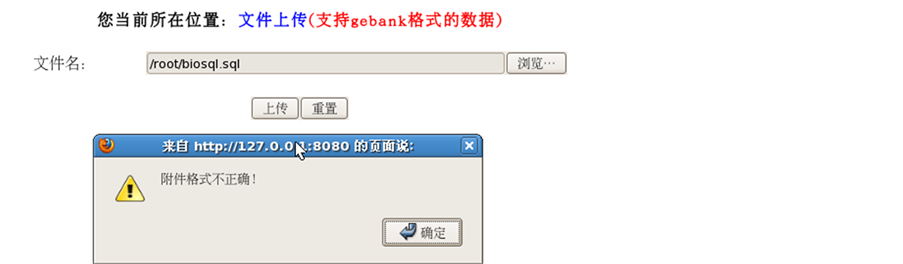
Figure 9. File upload
图9. 文件上传
4. 结束语
在生物技术时代,生物信息的研究与计算机技术的结合已是密不可分。建立生物分子二次数据库,可以针对生物学领域内的某一物种进行更深入的研究。且手工的记录和分析不在适应快速发展的生物基因信息的分析与使用。因此关于基因信息数据库及其平台的应用引起了广泛关注。Cynthia Gibas和Per Jambeck等通过对java、C、FORTRAN等内在程序语言编写的生物学软件调查后,认为perl是理想的选择,原因在于相比之下perl更有效率(efficiency)[12] 。而java作为一门编程语言,相对于其他语言,跨平台性好,可移植性强,入门简单。是目前编程人员的首选的语言。因此本平台结合perl的生物模块,即bioperl,和java语言在生物信息人员研究出高效的算法下(即将发表),实现了以抗逆基因二次数据库为例的构建与应用。此平台同样适用于其他本平台的分子生物的研究。不足之处,目前已满足相关功能,在后期通过提供blast序列比对接口和数据下载,插入许多数据处理应用,如数据的格式转换、可视化显示等完善本平台功能。
项目基金
自治区自然科学基金(批准号:2010211022)资助项目。新疆师范大学研究生科技创新基金资助项目(20131203)。
参考文献 (References)
- [1] 罗静初 (2000) 分子生物信息数据库. The 2nd Cross-Straits Symposium on Biology-Inspired Laboratory Workshop,中国高等科学技术中心, 41.
- [2] 邢仲璟, 林丕源, 林毅申 (2004) 基Bioperl的生物二次数据库建立及应用. 计算机系统应用, 11, 58-60.
- [3] 周猛, 童春发, 施季森 (2008) 充分利用Bioperl加速生物信息学的研究. 生物信息学, 1, 43-45.
- [4] 白琳 (2012) 植物抗逆基因资源平台的构建与分析. 浙江大学, 杭州.
- [5] BioPerl. Installation. http://www.bioperl.org/wiki/Installing_BioPerl
- [6] 孙啸, 陆祖宏, 谢建明 (2005) 生物信息学基础. 清华大学出版社, 北京.
- [7] 向福, 陈悟, 余龙江 (2004) 基于Bioperl的基因序列获取的程序设计与实现. 生物技术, 6, 64-66.
- [8] biosql操作指南. http://www.biosql.org/wiki/Main_Page
- [9] 郭文久 (2007) Perl语言环境下生物信息学的数据库技术. 安康学院学报, 5, 74-78.
- [10] 贾广宇 (2006) MVC设计模式下Web开发框架的研究与应用. 大连海事大学, 大连.
- [11] 宋利军 (2003) RedHat Linux 9.0实用教程. 科学出版社, 北京, 121-122.

NOTES
*通讯作者。