Open Journal of Nature Science
Vol.03 No.02(2015), Article ID:15280,6
pages
10.12677/OJNS.2015.32004
A Hypothesis: TGF-β1/Smad3 Signaling Pathway Participates in the Development of Parkinson’s Disease
Yongpeng Yu1,2
1Department of Neurology, Affiliated Wendeng Center Hospital of Weifang Medical College, Weihai Shandong
2The Medical College, Qingdao University, Qingdao Shandong
Email: yypeng6688@126.com
Received: May 5th, 2015; accepted: May 19th, 2015; published: May 26th, 2015
Copyright © 2015 by author and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



ABSTRACT
Parkinson’s disease (PD), which is one of neurodegenerative diseases, is a serious threat to human health. So far there has been no special treatment for it. PD onset is closely associated with the disorders of iron metabolism and its inducing and mediating oxidative stress response in the brain. The mechanism of its regulation is still elusive. Recently it was found that transforming growth factor-β1 (TGF-β1) can down-regulate the expression of ferritin heavy chain (FHC) and lead to cell labile iron increasing. It was found that TGF-β1/Smads signaling pathway could regulate cellular iron transport and metabolic balance by regulating hepcidin (Hep) expression in the hemochromatosis research. This review focused on the possible mechanism of TGF-β1/Smads signaling pathway involving iron metabolism and oxidative stress regulation and proposed a medical hypothesis: TGF-β1/Smad3 signaling pathway might participate in the process of PD development. It is expected that the experiment will be performed to explore the effect of abnormal regulation of this signaling pathway on the iron metabolism protein expressions and iron levels in PD, and to investigate regulatory mechanism of the TGF-β1 signaling on oxidative stress in dopaminergic neurons. It will be of great significance to reveal the mechanism of PD, and to find effective treatments for it.
Keywords:Parkinson’s Disease, Iron, TGF-β1, 6-Hydroxydopamine
假说:TGF-β1/Smad3信号通路参与帕金森病的发生发展
于永鹏1,2
1潍坊医学院附属文登中心医院神经内科,山东 威海
2青岛大学医学院,山东 青岛
Email: yypeng6688@126.com
收稿日期:2015年5月5日;录用日期:2015年5月19日;发布日期:2015年5月26日

摘 要
帕金森病(Parkinson’s disease, PD)是严重威胁人类健康的神经变性疾病之一,迄今没有特殊有效的治疗方法。脑铁代谢紊乱及其介导的氧化应激反应与PD发病关系密切,但其调节机制尚不明确。新近发现,转化生长因子(Transforming growth factor-β1, TGF-β1)能下调铁蛋白重链(Ferritin heavy chain, FHC)的表达并导致细胞易变铁库增加,在血色素沉着病研究中发现TGF-β1/Smads信号通路通过调节铁调素(Hepcidin, Hep)的表达调控细胞铁代谢。本文重点论述TGF-β1/Smads信号通路参与铁代谢及氧化应激调控的可能机制,并提出医学假说:TGF-β1/Smad3信号通路参与PD的发生发展过程。希望将来能以TGF-β1/Smad3信号通路为切入点,探讨该通路的异常调控对PD脑铁代谢相关蛋白表达及铁水平的影响,并探讨TGF-β1信号对多巴胺能神经元氧化应激调控的机制,这对揭示PD发病机制、寻找有效的治疗方法具有重要意义。
关键词 :帕金森病,铁,转化生长因子-β1,6-羟基多巴胺

1. 引言
帕金森病(Parkinson’s disease, PD)是严重威胁人类健康的神经变性疾病之一,迄今没有特殊有效的治疗方法。虽然左旋多巴是目前治疗PD最有效的药物,但也只能改善症状、并不能阻止疾病进展,且长期应用多巴制剂会出现疗效减退、运动障碍、症状波动和精神障碍等严重并发症。目前诸多神经保护性治疗措施逆转PD病程的能力尚需要进一步评估,至今对其神经保护作用仍存在较大争议。因此,阐明其发生发展机制是实现该目标必须解决的关键问题。转化生长因子β (Transforming growth factor-β, TGF-β)是一类具有多种生物学活性的多肽类细胞因子,除了TGF-β异构体之外,该家族还包括抑制素(Inhibins)、活化素(Activins)、骨形态发生蛋白(BMP)、生长分化因子(GDF)、Nodal、抗穆勒管激素(AMH)等近30种蛋白。这些生长因子对细胞增殖、分化和死亡,控制胚胎发育、组织分化和成体内器官组织的内平衡起重要的调节作用。研究发现TGF-β1通过调节铁蛋白重链(Ferritin heavy chain, FHC)的表达导致细胞易变铁库的增加,在促进肿瘤的发展中起重要作用[1] 。在血色素沉着病的研究中发现TGF-β1/Smads信号通路通过调节铁调素(Hepcidin, Hep)的表达调控细胞铁的转运和代谢平衡[2] 。研究发现TGF-β/Smads信号通路与细胞氧化应激关系密切[3] -[6] 。我们前期研究表明在6-羟基多巴胺(6-OHDA)损伤大鼠的黑质纹状体区氧化应激增强,主要表现为铜锌超氧化物歧化酶(CuZnSOD)、谷胱甘肽过氧化物酶(GSH-Px)、还原性谷胱甘肽(GSH)等抗氧化物质的活性和含量降低[7] ,推测在PD中TGF-β1/Smads信号通路可能参与铁代谢及其介导的氧化应激的调控。本文重点论述TGF-β1/Smads信号通路在铁代谢及氧化应激调控中的可能机制,并提出医学假说:TGF-β1/Smad3信号通路可能参与PD的发生发展过程。
2. 帕金森病与铁代谢
研究证实某些神经系统变性疾病(NDs)包括Halle-rvorden-spatz综合症(HSS)、PD、阿尔茨海默(AD)和亨廷顿舞蹈病(HD)与铁代谢内稳态失衡引起铁水平的增高有关,伴有转铁蛋白降低,同时氧化应激反应增强[8] 。在PD发生发展过程中,铁代谢失调被认为是PD的重要特征。铁代谢异常及其介导的氧化应激损伤是造成黑质多巴胺能神经元选择性损伤的主要机制之一[9] 。近来研究发现PD黑质存在更多的易变铁(Labile Iron, LI)[易变铁是指非铁蛋白结合的、参与氧化应激反应的二价或三价铁离子,又称易变铁库],PD黑质氧化应激损伤可能与超负荷的易变铁有关[10] 。最近某些脑铁代谢相关基因突变的发现强有力地支持:在某些NDs中脑铁的升高是神经元死亡的始发因素这一假设[11] 。在这些疾病中,脑铁代谢异常与铁代谢相关蛋白功能异常或协调表达调控异常密切相关。铁代谢相关蛋白或肽主要有铁蛋白(Ferritin)、转铁蛋白(Tf)、IRPs、黑素转铁蛋白(MTf)、HP、FP1、DMT1、Hep、十二指肠细胞色素B(Dcyth)等。其中除Ferritin不在星形胶质细胞表达外,TfR、Dcytb、DMT1、FP1、IRPs在脑内不同细胞(神经元、星形胶质细胞、小胶质细胞)均有表达。随着对胶质细胞研究的不断深入,发现其在神经生理和病理中有着重要作用,PD脑黑质致密部除了大量的多巴胺能神经元缺失外,还存在不同程度小胶质细胞和星形胶质细胞的激活,提示胶质细胞参与了PD的发病过程。研究发现不同类型的胶质细胞在铁的摄取、储存和转运中发挥的作用不同。小胶质细胞是脑内的巨噬细胞,主要捕获脑内的自由铁并储存于铁蛋白中。正常情况下,星形胶质细胞中没有铁积累,但在铁代谢异常时,星形胶质细胞也出现铁的积累。研究发现在6-OHDA诱导的PD模型大鼠黑质区FP1、Hp、IRPs、DMT1表达上调[12] -[14] 。虽然关于脑铁代谢的研究初步表明铁代谢相关蛋白表达异常与PD发病有关,但很多关于PD脑铁稳态及其调节机制的关键问题仍不清楚,尤其是对PD铁稳态的调节机制仍知之甚少,是需要将来进一步探讨的科学问题。铁及其介导的氧化应激与PD发病的关系如图1所示 [6] 。
3. TGF-β1/Smads信号通路与帕金森病
TGF-β家族有多种异构体,在哺乳动物体内有3种亚型:TGF-β1,TGF-β2,TGF-β3。Smad是TGF-β超家族信号传导的下游的因子,介导TGF-β信号由细胞质传入细胞核,特异性的调节TGF-β靶基因的表达。Smad在结构上含有高度保守的N端结构域(MH1)和C端结构域(MH2),根据Smad家族各个因子在TGF-β信号转导中的作用不同,分为受体激活型Smad (R-smad)、公用型Smad (Co-smad)和抑制型Smad (I-smad)。R-Smads,包括Smad1、2、3、5、8,是TGF-β1型受体激酶的底物,其中Smad2、3介导TGF-β1信号。Co-Smads,包括Smad4和爪蟾的Smad 4β (Smad10),它通过与R-Smads的结合参与信号传递。I-Smads,包括Smad6、7,其中Smad7抑制TGF-β1介导的R-Smads的信号传递。其中TGF-β1是公认最重要的调控因子,通过该信号通路的下游关键分子Smad2/3而发挥作用,Smad4在信号转导中起瓶颈作用。其中TGF-β1活化的I型受体TGF-βRI(ALK5)是TGF-β1信号转导的关键性节点,SB-431542 (4-(5-benzo [1, 3]dioxl-5 -yl 4-pyridin-2-yl-1 H-imidazol-2-y1)-benzamide)是特异性TGF-β1受体抑制剂,进而阻断Smad3蛋白的活化和入核,最终阻断TGF-β1/Smad3信号转导,TGF-β家族成员信号转导示意图见图2。
TGF-β首先与2型受体结合成复合物,TGFβR2二聚化后与TGF-β结合形成四聚体,同时TGF-β发

Figure 1. The relationship between iron and iron-mediated oxidative stress with the onset of PD [6]
图1. 铁及其介导的氧化应激与PD发病的关系[6]
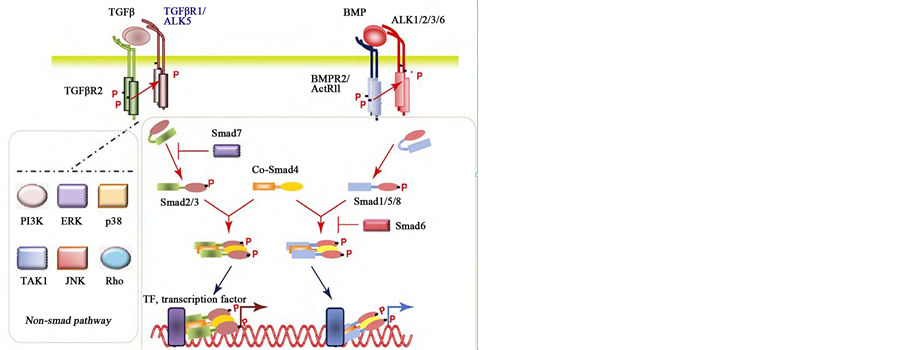
Figure 2. The diagram of TGF-β family signal transduction
图2. TGF-β家族成员信号转导示意图
生构系的改变,被TGFβR2所识别并结合,形成TGFβR2-TGFβ-TGFβR2复合物复合物中TGFβR2被TGFβR2磷酸化后激活TGFβR2,将信号转入细胞内,使Smads2、3蛋白中的丝氨酸磷酸化,此步骤可被Smad7抑制。磷酸化的Smad2/3蛋白与Smad4蛋白结合转运到细胞核从而调节靶基因的转录。同样BMP与其具有丝氨酸/苏氨酸激酶活性的II型受体(BMPR2和ActRIIB)结合,再招募I型受体(ALK3/BMPRIA、ALK6/BMPRIB和ALK2/ActRI)并使之磷酸化。磷酸化的BMPRI再招募效应分子Smad1/5/8 (R-Smads),并使R-Smads C末端磷酸化。磷酸化的R-Smads与Smad4结合并转运至核内,在其他转录因子的协同作用下调控靶基因的转录。
异常的TGF-β信号已经在很多疾病中被阐明,如肿瘤和纤维化。对于肿瘤,TGF-β表现出“双向”作用。即TGF-β信号能抑制肿瘤细胞生长,但TGF-β激活的信号具有促肿瘤的作用。TGF-β1及其受体在中枢神经系统广泛表达。近来研究发现TGF-β1在神经细胞中可能也存在“双向”作用。有研究显示在PD脑黑质区和脑脊液中TGF-β1表达增多[15] ,在AD患者脑脊液和血清中TGF-β1水平亦升高[16] 。同样在缺血性脑损伤、脊髓外伤、肌萎缩侧索硬化及脑积水患者神经组织中TGF-β1亦有类似的变化[17] -[20] 。有研究发现,腺病毒介导的TGF-β1过表达导致MPTP处理的小鼠黑质多巴胺能神经元存活下降同时伴随着纹状体多巴胺耗竭增加[21] 。Smad3基因敲出导致小鼠黑质多巴胺能神经元变性、星形胶质细胞数量减少并诱导α-synuclein蛋白的聚集[22] 。Smad3基因缺失减低小鼠脑神经前体细胞的迁移并影响神经的发生和发育[23] 。TGF-β-Smad2/3信号通路也参与了谷氨酸盐介导的运动神经元的损伤[24] 。然而亦有研究显示TGF-β1具有神经保护作用,TGF-β1在星形胶质细胞或神经元中过表达能够抵御神经损伤[25] 。在中枢神经系统,增加TGF-β1的表达水平可能作为对抗β淀粉样蛋白前体诱导的神经变性的一种措施[26] 。以上研究结果表明,在不同器官、组织和细胞中TGF-β1功能作用可能是多样化的。TGF-β1及Smad3可能在包括PD在内的神经变性疾病的发生发展过程中发挥某些重要的调控作用,Nox介导的TGF-β 诸多生物学效应见图3。
4. TGF-β1/Smads信号通路与铁代谢
近来研究发现除了BMP信号对铁代谢调节发挥重要作用外,TGF-β1信号通路与细胞铁代谢之间也存在密切的联系。最近研究表明TGF-β1下调FHC及其导致的易变铁库的增加在上皮细胞–间质细胞转化(EMT)过程中起关键作用[1] 。在血色素沉着病的研究中发现,TGF-β1/Smads信号通路通过调节Hep的表达调控细胞铁的转运和代谢平衡,肝细胞中Smad4缺乏能降低Hep的表达并导致铁在多器官中沉积[2] 。铁能抑制肾小管上皮细胞TGF-β1的产生并延缓肾损伤的修复[27] 。在H-ras恶性转化的纤维肉瘤中,TGF-β1能够选择性的调控Ferritin的表达[28] 。在软骨细胞分化过程中,乳铁蛋白表达受TGF-β1下游胞内信号Smad3的调控[29] ,提示TGF-β1信号通路参与细胞铁代谢的调节。最近研究发现,在星形胶质细胞中TGF-β1上调FP1的表达,但对DMT1表达没有影响。然而在小胶质细胞,TGF-β1却诱导DMT1和抑制FP1的表达,提示TGF-β1在星形胶质细胞和小胶质细胞铁稳态调节中的作用是不同的[30] 。鉴于TGF-β1信号与铁蛋白表达及铁代谢之间存在密切联系,推测TGF-β1信号可能参与铁代谢相关蛋白的表达及细胞铁稳态的调控,从而影响PD的起始和进展。
5. TGF-β1/Smads信号通路与氧化应激
细胞内活性氧产物(ROS)与TGF-β应答基因关系密切。除了Smads,MAPK通路(包括:JNK、p38、ERK-MAPK和PI3K-Akt)也对TGF-β1信号转导产生影响。MAPK信号途径能调控ROS参与的TGF-β纤维化效应。研究显示,在肾小管上皮细胞中TGF-β1可明显上调NADPH氧化酶(Nox)表达,并引起细胞内ROS增加[31] 。Nox4是NADPH氧化酶同源物Nox家族中的一员,能够透过质膜传递电子产生ROS,是公认的能够调节ROS产生的特殊功能酶。在体内外多种细胞中TGF-β1通过介导Nox4刺激ROS的产生,TGF-β1介导的ROS的增加是纤维化发病机制的主要特征。研究发现Smads信号通路受细胞氧化还

Figure 3. Multiple biological effects of Nox-mediated TGF-β
图3. Nox介导的TGF-β诸多生物学效应
原状态的影响,Smad3基因敲出小鼠黑质DA代谢及氧化应激增加[22] 。在气道平滑肌细胞中,TGF-β1能够诱导Nox4的表达,同时抑制锰过氧化物酶(MnSOD)和过氧化氢酶(CAT)的表达[32] 。免疫组化和原位杂交显示Nox4在神经元中表达。抑制Nox4的表达能阻止缺血性卒中后神经组织的氧化应激反应和神经变性[33] 。以上结果提示TGF-β1与细胞氧化应激关系密切,并且Nox4是其重要的下游调节信号,Nox介导的TGF-β诸多生物学效应见图3。
6. 小结
综上所述,TGF-β1/Smads、铁代谢、氧化应激与PD发病关系密切。既往虽然对TGF-β1/Smads信号通路进行了大量的研究,然而关于该信号通路在PD脑铁代谢和氧化应激调控作用及其分子机制仍知之甚少,鉴于TGF-β1/Smad3信号通路参与铁代谢和氧化应激调控在其他诸多疾病发生发展中发挥重要作用,我们推测TGF-β1/Smad3信号通路可能也参与PD的发生发展进程。希望将来能以TGF-β1/Smad3信号通路为切入点,探讨PD中该通路的异常调控对不同细胞铁代谢相关蛋白表达及铁水平的影响,并探讨TGF-β1信号对多巴胺能神经元氧化应激调控的机制,这对揭示PD发病机制、寻找有效的治疗方法具有重要意义。
基金项目
本研究受国家自然科学基金项目资助(81400957)和威海市科技发展计划项目资助(2011-2-91-2)。
文章引用
于永鹏, (2015) 假说:TGF-β1/Smad3信号通路参与帕金森病的发生发展
A Hypothesis: TGF-β1/Smad3 Signaling Pathway Participates in the Development of Parkinson’s Disease. 自然科学,02,19-25. doi: 10.12677/OJNS.2015.32004
参考文献 (References)
- 1. Zhang, K.H., Tian, H.Y., Gao, X., et al. (2009) Ferritin heavy chain-mediated iron homeostasis and subsequent increased reactive oxygen species production are essential for epithelial-mesenchymal transition. Cancer Research, 69, 5340-5348.
- 2. Milward, E., Johnstone, D., Trinder, D., et al. (2007) The nexus of iron and inflammation in hepcidin regulation: SMADs, STATs, and ECSIT. Hepatology, 45, 253-256.
- 3. Yang, W.H., Deng, Y.T., Hsieh, Y.P., Wu, K.J. and Kuo, M.Y. (2015) NADPH oxidase 4 mediates TGFβ1-induced CCN2 in gingival fibroblasts. Journal of Dental Research, pii: 0022034515580986.
- 4. Oruqaj, G., Karnati, S., Vijayan, V., Kotarkonda, L.K., Boateng, E., Zhang, W., Ruppert, C., Günther, A., Shi, W, and Baumgart-Vogt, E. (2015) Compromised peroxisomes in idiopathic pulmonary fibrosis, a vicious cycle inducing a higher fibrotic response via TGF-β signaling. Proceedings of the National Academy of Sciences of the United States of America, 112, E2048-2057.
- 5. Yang, Y., Kim, B., Park, Y.K., Koo, S.I. and Lee, J.Y. (2015) Astaxanthin prevents TGFβ1-induced pro-fibrogenic gene expression by inhibiting Smad3 activation in hepatic stellate cells. Biochimica et Biophysica Acta (BBA)—General Subjects, 1850, 178-185.
- 6. Liu, R.M. and Gaston Pravia, K.A. (2010) Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radical Biology and Medicine, 48, 1-15.
- 7. Yu, Y.P., Ju, W.P., Li, Z.G., et al. (2010) Acupuncture inhibits oxidative stress and rotational behavior in 6-hydroxydopamine lesioned rat. Brain Research, 1336, 58-65.
- 8. Zecca, L., Youdim, M.B., Riederer, P., Connor, J.R. and Crichton, R.R. (2004) Iron, brain ageing and neurodegenerative disorders. Nature Reviews Neuroscience, 5, 863-873.
- 9. Vila, M. and Przedborski, S. (2004) Genetic clues to the pathogenesis of Parkinson’s disease. Nature Medicine, 10, S58-S62.
- 10. Wypijewska, A., Galazka-Friedman, J., Bauminger, E.R., Wszolek, Z.K., Schweitzer, K.J., Dickson, D.W., Jaklewicz, A., Elbaum, D. and Friedman, A. (2010) Iron and reactive oxygen species activity in parkinsonian substantia nigra. Parkinsonism & Related Disorders, 16, 329-333.
- 11. Ke, Y. and Ming, Q.Z. (2003) Iron misregulation in the brain: A primary cause of neurodegenerative disorders. The Lancet Neurology, 2, 246-253.
- 12. Song, N., Wang, J., Jiang, H. and Xie, J. (2010) Ferroportin 1 but not hephaestin contributes to iron accumulation in a cell model of Parkinson’s disease. Free Radical Biology & Medicine, 48, 332-341.
- 13. Jiang, H., Song, N., Xu, H., Zhang, S., Wang, J. and Xie, J. (2010) Up-regulation of divalent metal transporter 1 in 6-hydroxydopamine intoxication is IRE/IRP dependent. Cell Research, 20, 345-356.
- 14. Wang, J., Jiang, H. and Xie, J.X. (2007) Ferroportin1 and hephaestin are involved in the nigral iron accumulation of 6-OHDA-lesioned rats. European Journal of Neuroscience, 25, 2766-2772.
- 15. Nagatsu, T., Mogi, M., Ichinose, H. and Togari, A. (2000) Changes in cytokines and neurotrophins in Parkinson’s disease. Journal of Neural Transmission. Supplementum, 60, 277-290.
- 16. Rota, E., Bellone, G., Rocca, P., Bergamasco, B., Emanuelli, G. and Ferrero, P. (2006) Increased intrathecal TGF-beta1, but not IL-12, IFN-gamma and IL-10 levels in Alzheimer’s disease patients. Neurological Sciences, 27, 33-39.
- 17. Buss, A., Pech, K., Kakulas, B.A., Martin, D., Schoenen, J., Noth, J. and Brook, G.A. (2008) TGF-beta1 and TGF- beta2 expression after traumatic human spinal cord injury. Spinal Cord, 46, 364-371.
- 18. Ilzecka, J., Stelmasiak, Z. and Dobosz, B. (2002) Transforming growth factor-beta 1 (TGF-beta 1) in patients with amyotrophic lateral sclerosis. Cytokine, 20, 239-243.
- 19. Krupinski, J., Kumar, P., Kumar, S. and Kaluza, J. (1996) Increased expression of TGF-beta 1 in brain tissue after ischemic stroke in humans. Stroke, 27, 852-857.
- 20. Li, X., Miyajima, M., Jiang, C. and Arai, H. (2007) Expression of TGF-betas and TGF-beta type II receptor in cerebrospinal fluid of patients with idiopathic normal pressure hydrocephalus. Neuroscience Letters, 413, 141-144.
- 21. Sánchez-Capelo, A., Colin, P., Guibert, B., Biguet, N.F. and Mallet, J. (2003) Transforming growth factor beta1 overexpression in the nigrostriatal system increases the dopaminergic deficit of MPTP mice. Molecular and Cellular Neuroscience, 23, 614-625.
- 22. Tapia-González, S., Giráldez-Pérez, R.M., Cuartero, M.I., Casarejos, M.J., Mena, M.Á., Wang, X.F. and Sánchez- Capelo, A. (2011) Dopamine and α-synuclein dysfunction in Smad3 null mice. Molecular Neurodegeneration, 6, 72.
- 23. Wang, Y. and Symes, A.J. (2010) Smad3 deficiency reduces neurogenesis in adult mice. Journal of Molecular Neuroscience, 41, 383-396.
- 24. Katsuno, M., Adachi, H., Banno, H., Suzuki, K., Tanaka, F. and Sobue, G. (2011) Transforming growth factor-β signaling in motor neuron diseases. Current Molecular Medicine, 11, 48-56.
- 25. Wyss-Coray, T. (2006) TGF-Beta pathway as a potential target in neurodegeneration and Alzheimer’s. Current Alzheimer Research, 3, 191-195.
- 26. Caraci, F., Battaglia, G., Bruno, V., Bosco, P., Carbonaro, V., Giuffrida, M.L., Drago, F., Sortino, M.A., Nicoletti, F. and Copani, A. (2011) TGF-β1 pathway as a new target for neuroprotection in Alzheimer’s disease. CNS Neuroscience & Therapeutics, 17, 237-249.
- 27. Horino, T., Ito, H., Yamaguchi, T., Furihata, M. and Hashimoto, K. (2005) Suppressive effects of iron on TGF-beta1 production by renal proximal tubular epithelial cells. Nephron Experimental Nephrology, 100, e1-e10.
- 28. Lo, J. and Hurta, R.A. (2000) Transforming growth factor beta1 selectively regulates ferritin gene expression in malignant H-ras-transformed fibrosarcoma cell lines. Biochemistry and Cell Biology, 78, 527-535.
- 29. Takayama, Y. and Mizumachi, K. (2010) Inhibitory effect of lactoferrin on hypertrophic differentiation of ATDC5 mouse chondroprogenitor cells. BioMetals, 23, 477-484.
- 30. Rathore, K.I., Redensek, A. and David, S. (2012) Iron homeostasis in astrocytes and microglia is differentially regulated by TNF-α and TGF-β1. Glia, 60, 738-750.
- 31. Zhang, H., Jiang, Z., Chang, J., Li, X., Zhu, H., Lan, H.Y., Zhou, S.F. and Yu, X. (2009) Role of NAD(P)H oxidase in transforming growth factor-beta1-induced monocyte chemoattractant protein-1 and interleukin-6 expression in rat renal tubular epithelial cells. Nephrology (Carlton), 14, 302-310.
- 32. Michaeloudes, C., Sukkar, M.B., Khorasani, N.M., Bhavsar, P.K. and Chung, K.F. (2011) TGF-β regulates Nox4, MnSOD and catalase expression, and IL-6 release in airway smooth muscle cells. AJP: Lung Cellular and Molecular Physiology, 300, L295-L304.
- 33. Kleinschnitz, C., Grund, H., Wingler, K., Armitage, M.E., Jones, E., Mittal, M., Barit, D., Schwarz, T., Geis, C., Kraft, P., Barthel, K., Schuhmann, M.K., Herrmann, A.M., Meuth, S.G., Stoll, G., Meurer, S., Schrewe, A., Becker, L., Gailus-Durner, V., Fuchs, H., de Klopstock, T., Angelis, M.H., Jandeleit-Dahm, K., Shah, A.M., Weissmann, N. and Schmidt, H.H. (2010) Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biology, 8, e1000479.