Interdisciplinary Science Letters
Vol.
02
No.
04
(
2018
), Article ID:
28686
,
21
pages
10.12677/ISL.2018.24021
Progress in Computational Chemistry
Ning Qin1,2, Qing Min1*, Bo Li3, Mixia Ma3, Kaiyuan Shao2, Wenxiang Hu2,3,4*
1School of Pharmacy, Hubei University of Science and Technology, Xianning Hubei
2Jingdong Xianghu Microwave Chemistry Union Laboratory, Beijing Excalibur Space Military Academy of Medical Sciences, Beijing
3School of Chemistry and Environmental Engineering, Wuhan Institute of Technology, Wuhan Hubei
4Space Systems Division, Strategic Support Troops, Chinese People’s Liberation Army, Beijing
Received: Jan. 2nd, 2019; accepted: Jan. 22nd, 2019; published: Jan. 29th, 2019

ABSTRACT
This paper describes the concepts of computational chemistry, calculation methods and application fields. It discusses its application in the auxiliary molecular design and structure-activity relationship of drugs and its auxiliary material design and synthetic route design, and it looks forward to its application prospects in the field of chemistry and virtual reality.
Keywords:Computational Chemistry, Calculation Method, Molecular Structure, Molecular Simulation, Structure-Activity Relationship, Virtual Reality
计算化学相关研究进展
秦宁1,2,闵 清1*,李博3,马密霞3,邵开元2,胡文祥2,3,4*
1湖北科技学院药学院,湖北 咸宁
2北京神剑天军医学科学院京东祥鹄微波化学联合实验室,北京
3武汉工程大学,化学与环境工程学院,湖北 武汉
4中国人民解放军战略支援部队航天系统部,北京

收稿日期:2019年1月2日;录用日期:2019年1月22日;发布日期:2019年1月29日

摘 要
本文叙述了计算化学概念、计算方法及其在辅助药物分子设计、辅助材料设计和合成路线设计等方面的应用,展望了计算化学在化学相关领域以及虚拟现实等方面的应用前景。
关键词 :计算化学,计算方法,分子结构,分子模拟,构效关系,虚拟现实

Copyright © 2018 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 前言
随着科学技术的发展,化学家们从利用塑料短杆和小球表示分子结构发展到使用计算机来展示各种化学模型。由此,产生一门新兴的交叉学科,即计算化学。所谓计算化学(computational chemistry),是一门应用计算机研究化学反应和物质变化的科学,已经成为理论化学的一个重要分支。它是利用有效的数学近似以及计算机程序计算分子的性质,包括电子、轨道参数和几何参数等,用以解释一些具体的化学问题,在电子、原子、分子水平上阐明化学现象和化学问题的本质,包括反应机理、化学性质及药物的构效关系等。
二十世纪八十年代以来,先进的分析仪器的应用、量子化学计算方法和计算机技术的飞速发展,对化学科学的发展产生了革命性的影响。其研究内容、方法乃至学科的结构和性质都在发生深刻的变化。
化学的发展历程是人类运用了认识自然的两种科学方法:归纳法和演绎法。归纳法也叫归纳推理法,它是一种由个别性知识推出一般性结论的推理过程。演绎法也叫演绎推理法。它是由一般性的前提出发,通过推导即“演绎”,得出具体陈述或个别结论的过程。
这两种认知的科学方法是互相依赖、互为补充,紧密联系的。演绎推理的一般性知识(大前提),来源于归纳推理的概括和总结;而单靠归纳推理(除了完全归纳)是不能证明必然性,并且归纳的分析、综合过程需要借助于理论思维,依靠人们先前积累的一般性理论知识的指导。因此,在归纳推理的过程中,人们常常需要应用演绎推理对某些归纳的前提或者结论加以论证。只有两者结合应用,才能认识事物的本质及其发展规律。
但是,化学长期以来一直被科学界公认为一门纯实验科学,是基于实验事实为依据的科学。即化学长期采用归纳法作为研究方法,而演绎法在化学界尚未获得广泛应用。究其原因主要是研究的对象太复杂加上传统观念根深蒂固。直到上世纪80年代,这种境况有所改善。
二十世纪二、三十年代诞生的计算化学迎合了时代发展的需要,在七十年代和八十年代得到了较大发展,至九十年代它已完全成为一门独立的学科,受到了国际化学界的广泛重视,而计算量子化学在1998年获得诺贝尔化学奖,确立了计算化学在化学和整个自然科学的重要地位。瑞典皇家科学院在颁奖公报中宣告“…量子化学已发展成为广大化学家都能使用的工具,将化学带入一个新时代—实验与理论能携手协力揭示分子体系的性质。化学不再是一门纯实验科学了”。它是与化学、数学、计算机科学、物理学、药物学、材料科学等学科高度交叉、相互渗透的新的生长点,是许多实用技术的基础,并深受当今计算机与网络通讯技术飞速发展的影响,而处在迅速发展和不断演变之中。可以预见,计算机化学将成为化学发展的“领头羊”之一。
2. 计算化学与诺贝尔化学奖
马丁·卡普拉斯(Martin Karplus)、迈克尔·莱维特(Michael Levitt)和亚利耶·瓦谢尔(Arieh Warshel)三位科学家因在“发展复杂化学体系多尺度模型”计算化学方面所做出的重要贡献而获得2013年诺贝尔化学奖。Martin Karplus于1930年在奥地利维也纳出生,1953年获得美国加州理工大学博士学位,现任法国斯特拉斯堡大学及美国哈佛大学教授。Michael Levitt于1947年在南非勒陀利亚出生,1971年获得英国剑桥大学博士学位,现任美国斯坦福大学医学院教授。Arieh Warshel于1940年在以色列Kibbutz Sde-Nahum,1969年获得以色列魏茨曼科学研究所博士学位,现在是美国南加州大学杰出教授。在上世纪70年代,Martin Karplus, Michael Levitt和Arieh Warshel的一系列研究工作为化学反应模型的应用奠定了理论基础,有利于我们对化学反应过程的理解与预测。时至今日,化学领域所取得的大部分重要进展都离不开先进计算机模型的帮助。
1998年,沃尔特·库恩(Walter Kohn)和约翰·波普尔(John Pople)两位理论化学家因“有效地发展了计算方法和计算程序,将量子力学的方程用于解决化学问题”而获得了诺贝尔化学奖。在前者工作的基础上,Karplus,Levitt和Warshel成功地将牛顿的经典物理学工作与性质上完全不同的量子物理学相互协调。经典物理的强大之处在于其计算过程相对简单,并且可以拥有模拟非常大型的分子结构,但是它无法模拟化学反应过程。因此,科学家们将量子物理学合适的运用到模拟化学反应过程。三位科学家结合经典物理学和量子物理学,设计出多尺度复杂化学系统模型,将传统的化学实验搬到了网络世界。这一完美结合现实与理论的化学系统模型,为更全面了解并预测化学反应进程奠定了基础。
计算化学(computational chemistry)是近几十年中发展最快的化学研究领域之一,其在多种化学研究中都有广泛的应用。计算化学是根据基本的物理化学理论(通常指量子化学、统计热力学及经典力学)和大量的数值运算方式研究分子、团簇的性质及化学反应的一门科学 [1] [2] [3] [4] 。以量子化学理论和计算、分子反应动力学理论和计算、分子力学及分子动力学理论和计算等来探明相关的化学反应 [5] [6] 。此外,计算化学还可以通过理论计算等方式预测化学研究的方向等。计算化学发展至今,已经形成了独具特色的解决多种化学问题的方法和程序,例如:Gaussian09、Hyper Chem、ADF2004等专业软件的开发和不断的更新 [7] [8] ,大力推动了这门新兴的交叉学科的蓬勃发展。
3. 计算化学原理与方法
了解分子的物理与化学性质,才能知道它与其它分子的作用情况,才能知道由它组成的聚集体的某种性能,如分子及分子聚集体运动状态、势能、光、电、磁、热、微波等效应。要知道化学分子的性质,首先要知道分子的所有结构信息。因为分子结构中蕴含着大量的信息,包括组成它原子之间的连接情况、三维空间的构象、构型、电子运动、原子之间的相互作用、电子之间的相互作用、电子与原子核之间的相互作用、分子的能量等等。所有这些都需要通过实验测定和计算化学方法获取。
计算化学方法包括:计算方法和分子模拟。计算方法包括分子力学(MM)和量子力学(QM)方法;而分子模拟包括分子动力学(MD模拟)和统计力学(SM)模拟。
分子力学方法即是经典的牛顿力学方法。即采用经典的物理定律预测分子结构和性质。它基于下述几点假设:① 原子核运动和电子的运动可以看成是独立的(Born-Oppenheimer近似);② 分子被看成一种由简单的元素像球(原子),棒(化学键)和柔性连接(键角和扭转角)的机械组件构成;③ 分子看作是由一个简谐力把原子结合维系在一起的集合;④ 这些简谐力由各个势函数来描述;⑤ 分子的总势能或空间位阻能是组成它的势函数的总和。
分子力学采用经典物理对分子进行处理,本质上说是能量最小化法,即在原子间相互作用势的作用下,通过粒子分布的几何构型,以能量最小为判据,从而获得体系的最佳结构。它是以分子间作用能、氢键作用能、偶极力作用能、扭矩作用能和键长、键角的变形能等来计算分子的能量,优化分子的空间构型。分子能量的基本构成为:
所以,分子力场的势函数基本构成为:
分子力场是描述各种形式的相互作用力对分子势能影响的势能函数,包括键合能和非键合能相互作用,键合能包括:键长的伸缩能、键角的弯曲能、二面角扭转能;非键能包括分子间作用能(范德华力能)、静电相互作用能、氢键能。分子力场的性能取决于势能函数和结构参数,其结构参数来源于大量的热力学和光谱学实验,或者来源于量子化学计算结果。到目前为止,分子力场总共有十多种,不同的分子力场,势能函数表达式有所不同,适合于研究不同的分子体系。每个分子力场,键合能部分基本相似,主要差别是非键合能部分。依据研究的对象选择不同的分子力场。
最主要的分子力场:
AMBER力场。它是传统力场之一。主要适用于蛋白质和核酸体系、多糖,最新的也可用于有机小分子。
CHARMm力场(Chemistry at Harvard Macromolecular mechanics)。也是传统力场之一。适用于各种分子性质的计算和模拟。对于从孤立的小分子到溶剂化的生物大分子体系的多种模拟体系都可以给出较好的结果,但不适合于有机金属配合物。
CVFF力场(Consistent Valence Force Field),属传统力场。适应于有机小分子和蛋白质体系。扩展后可用于某些无机体系的模拟,如硅酸盐、铝硅酸盐、磷硅化合物等,主要用于预测分子的结构和结合自由能。
MMX力场包括MM2和MM3,是目前应用最为广泛的力场,主要针对有机小分子。也可用于生物大分子体系,但计算速度较慢。
第二代力场比传统力场要更加复杂,涉及的力场参数更多,计算量也更大,当然也相应地更加准确。
CFF力场,属于第二代力场。CFF91主要用于模拟有机小分子、蛋白质以及小分子-蛋白质之间的相互作用。CFF95,除了CFF91功能外,还可用于高分子体系的模拟。而PCFF在91的基础上,还适用于聚碳酸酯、三聚氰胺甲醛树脂、多糖、核酸、分子筛等其它无机和有机材料体系的模拟。
MMFF94力场(Merk Molecular Force Field),属于第二代力场。它是目前最准确的力场之一,定义了非常完善的原子类型,既适应于有机小分子,也适用于大分子体系。
COMPASS力场(Condensed-phase Optimized Molecular Potentials for Atomistic Simulation Studies),属于第二代力场。用于原子水平模拟研究的凝聚态优化的分子力场,是第一个能同时预测气态和凝聚态性质的从头力场,适用于常见的有机小分子、无机小分子和高分子,也使用于金属、金属氧化合物及金属卤化物的模拟,较准确地预测晶体的多种性质。
此外,还有一些通用力场,如:ESFF力场(Extensive Systematic Force Field)用于有机分子、无机分子及有机金属化合物的结构预测。UFF力场(Universall Force Field)覆盖了周期表中所有元素,应用最为广泛。Dreiding力场适用于有机分子、生物大分子即位于主族的无机分子。通用力场是基于规则的力场(Rule-based Force Fields),具有较好的可移植性,较广的使用范围,但精度较差。
分子力学计算,并不计算电子相互作用,它是对分子结构的一种简化模型。分子力场函数为来自实验结果的经验公式,对分子能量的模拟比较粗糙,但与量子力学相比,计算量要小数十倍,所以计算速度也就快数十倍。在适当的范围内,分子力学计算的精度与量子力学计算相差无几。这对于大分子复杂体系,显然分子力学方法更有优势。如对于蛋白质体系,能够进行能量优化、结合常数计算、蛋白质折叠的动力学模拟、活性位点检测、结合位点设计等等。
分子力场的不足是:不能描述电子的跃迁(包括质子的吸附),不能描述电子转移现象,不能描述质子的传递(如酸碱反应),不能处理电子效应占主导的化学问题,如化学键的形成和断裂等。
量子力学(QM)方法。量子化学是应用量子力学的规律和方法来研究化学问题的一门学科。研究范围包括稳定和不稳定分子的结构、性能及其结构与性能之间的关系;分子与分子之间的相互作用;分子与分子之间的相互碰撞和相互反应等问题。微观粒子具有波粒二象性特征和遵从量子力学测不准原理(粒子的位置和动量不能同时有确定值),不符合描述宏观物质运动规律的牛顿运动力学定律。微观粒子或体系的性质,由状态波函数 唯一确定,服从薛定谔方程(Schrödinger equation)。这是迄今为止人类社会最伟大的10个公式之一,它揭示了微观粒子运动规律。
薛定谔方程分为含时和不含时两种:
其中, 是表征波函数总能量的哈密顿算符 , 是物理系统的波函数,i是虚数单位,h是约化普朗克常数, 是对于时间t的偏微分。方程在<10−13 m的微观层次。然而,
方程建立容易,但求解十分困难。
而不含时的薛定谔方程为:
;
其中, 是不含时波函数,E是能量。在三维空间里,处于位势 的单独粒子,其不含时薛定谔方程(即定态薛定谔方程)可以更具体地表示为:
其中,m是质量, 是拉普拉斯算符, 是不含时波函数,E是能量。
利用应用分离变量法,可以将不含时的转化为含时的。
不含时的薛定谔方程,即波函数与时间无关,是一种定态的方式,不依于时间,又称为本征能量薛定谔方程,或定态薛定谔方程。顾名思义,本征能量薛定谔方程,可以用来计算粒子的本征能量与其它相关的量子性质。
薛定谔方程是量子力学的基本方程,它揭示了微观物理世界物质运动的基本规律,就像牛顿定律在经典力学中所起的作用一样,它是原子物理学中处理一切非相对论问题的有力工具。薛定谔方程中,涵盖着分子的所有信息。所以,量子化学计算的基础就是求解电子运动的薛定谔方程,从中获得原子和分子的核外电子运动情况,进一步了解分子的结构、电荷分布,原子间结合能,结构与性质的关系,一直到反应途径(核运动规律)的研究。但是,由于体系的哈密顿算符比较复杂,薛定谔方程能够严格求解的情况寥寥可数,因此,需要引入各种近似方法来求解薛定谔方程。
量子化学计算是基于定态薛定谔方程和建立非相对论近似、Born-Oppenheimer近似和单电子近似的三个近似基础上,采用如微扰论、变分法、半经验近似、自洽场理论等求解。不同的近似方法有不同的适用范围。
非相对论近似:因为相对论效应是关于粒子能量与粒子质量和运动速率的关系。而非相对论近似是指不考虑任何相对论效应。即忽略了质量与速度的相对论效应的情况下,认定电子与原子核间只存在库仑力。然而,当电子运动速度接近光速时,这样的近似是不合适的。特别是对于重元素中的电子,由于受到质量大的原子核的影响,s轨道电子和p轨道电子的运动速度接近光速,而相对论效应对d轨道和f轨道的间接影响也十分显著。但对于绝大多数有机分子来说,非相对论近似是可行的,避免了方程算符中的相对论效应项,使得求解简单化。
Born-Oppenheimer近似:原子核的质量是电子质量的103~105倍,所以体系中电子的运动速度比原子核快得多。电子处于高速运动中,而原子核只是在它们的平衡位置附近热振动。但当核的位置发生微小变化时,电子能迅速调整自己的运动状态使之与变化后的库仑场相适应。换句话说,电子能绝热于原子核的运动。可以认为,当原子核位置发生微小变动时,迅速运动的电子可瞬时调整,达到新的平衡。即电子不会因原子核的运动而产生跃迁,只会引起各电子态连续的、绝热的变化,这就是所谓的Born-Oppenheimer近似或称绝热近似。
单电子近似:假定把每一个电子所受其它电子的库仑作用,近似的处理成一个平均的等效势场,这样就可以把多电子问题简化成单电子问题,这种近似称为单电子近似,也称为平均场近似。显然这种近似比较粗糙的,因为它忽略了电子之间的相互作用。因此,单电子近似过于粗糙,需要进行必要的修正,也因此引出了各种不同的算法。
根据上述三个近似,意味着在任意确定的核分布形式下,电子都有相应的运动状态;同时核间的相对运动可视为所有电子运动的平均结果。所以电子的波函数只依赖于原子核的位置,而不是他们的动能。于是近似认为,电子运动与原子核运动可以分开处理,可以将上式分解为:
电子运动方程:
原子核运动方程:
电子运动波函数
核运动波函数
电子运动方程中的Ee (势能面)仅仅与原子核坐标有关。相应的,核运动方程所表示的是在核势能面E(R)上的核运动方程。
求解电子运动波函数方程的量子化学计算,是把电子的波函数和能量处理成原子核坐标的函数。由于量子化学求解电子波函数和势能面耗时巨大,常常将势能面进行经验性的拟合,成为力场,由此构成分子力学的基础。将核运动方程用牛顿运动方程代替,势能面采用力场拟合,就构成了分子动力学基础。
3.1. 量子化学计算方法
3.1.1. 从头计算法
从头计算法是一种基于量子化学的第一性原理(First_Principles)使用严密的近似求解定态薛定谔方程的方法。它只采用5个基本物理常数:电子质量μ0、电子电量e、普郎克常数h、光速c、玻耳兹曼常数k,而不依赖任何经验参数即可合理地预测微观体系的状态和性质。在从头算法框架内,有两个求解方程的策略:基于波函数方法和基于密度泛函方法。
基于波函数方法,即以获得体系的波函数,来描述分子的结构和性质。最经典的是Hartree-Fock理论(HF理论)。它是采用单电子近似,总能量由分子轨道函数来表达,通过Slater行列式的线性耦合来描述波函数。但对于多原子的分子体系,Roothaan将分子轨道用原子轨道(或某些基组)展开,分子轨道成为原子轨道的线性组合(简称LCAO),这就形成了Hartree-Fock-Roothaan方程,简称HFR方程。HF理论最大问题是忽略了电子相关性。所以,后来发展了组态相互作用方法(CI法),基于多体微扰理论的MP法以及通过指数形式的耦合算符运算的耦合簇法(CC),即所谓的“后Hartree-Fock方法”,广泛应用于研究分子的电子结构的各方面性质。如平衡几何构型、电荷密度分布、键级分析、偶极矩、内旋转和翻转势垒、力常数、势能面、电离势及各种能谱等等。它的应用范围不断扩大,从静态性质到动态性质,从分子内到分子间相互作用,计算精度也逐步提高,成为计算化学最精确的手段。目前,最常用的是MP系列,包括MP2、MP4。但它们对于较大的分子体系,会造成巨大的计算成本。
基于密度泛函方法,即以电子密度函数的方法,来描述分子的结构和性质。密度泛函理论(DFT)方法是近年来比较流行的一种处理相互作用多粒子体系的量子化学近似计算方法。通过对电子动能和势能的平均化处理,借助变分法或数值方法,可以得到薛定谔方程的近似解,大大简化了电子结构的计算,且不受自旋污染(在非限制性计算中,可能存在其它自旋态对方程的求解而产生的误差)的影响。
密度泛函理论(DFT)使用电子密度而不是波函数来表述体系能量。在DFT计算中,哈密顿量的一项,交换-相关泛函,采用近似形式。
密度泛函理论的基础:
Thomas-Fermi近似:不用体系的波函数,而是用比较简单的单电子密度来求解薛定谔方程;对于多原子电子,其能量泛函,只考虑核与电子以及电子间的相互作用。
Hohenberg-Kohn定理:
定理一:对于基态分子,其电子能量和其他电子性质由其电子密度唯一确定。或者说体系的基态能量仅仅是电子密度的泛函。
定理二:以基态密度为变量,将体系能量最小化之后就得到了基态能量。
这两个定理意味着应用分子的电子密度可以计算基态分子的基态能量。电子密度则由方程的本征函数—KS轨道的自洽求解KS方程求得。
其中:
动能项:
电子与核的势能项:
电子与电子之间的作用能项:
交换相关能项: ,即为交换能和相关能之和。
Kohn-Sham方法:在Thomas-Fermi近似和Hohenberg-Kohn定理的基础上,运用传统的平均势理论来解决电子的相关问题,即引入局域密度近似(local-density approximation, LDA),导出了著名的科恩-沈(Kohn-Sham)方程:
其中:
而 为外势, 则为交换相关势能。
式中 为Kohn-Sham轨道 的轨道能。含有N个粒子的Kohn-Sham系统的电子密度则由下式给出:
DFT既克服了传统的ab initio HF SCF方法难以考虑电子相关作用的缺点,又避开了MP、CISD、CCSD等方法处理较大体系耗费时间的不足。因此,DFT为研究较大的体系提供了一条可能的途径。
在经过对密度泛函理论的各种修正和扩充后,可以得到相当准确的体系各种性质。基于波函数的方法,对于多电子体系,函数变量随着电子数的增加而呈指数级增加,如N个电子,就有3N个变量(每个电子有三个自由度);而对于基于密度泛函方法,只有3个变量,大大地简化计算过程,从而使得从头计算能力从几十个原子迅速扩大到上千个原子。因此,DFT为研究较大的体系提供了一条可能的途径,广泛地用于化学反应和晶体结构的计算中。目前,密度泛函理论获得迅速扩展。密度泛函的基态能量是由电子动能、电子对核的势能、电子与电子之间的作用能以及交换相关能组成。前三项是严格按照本征方程求解,不含近似成分,而最后一项的交换相关能泛函无法精确求解,只能近似求解。所以,DFT方法的精确度集中在交换相关能泛函上,也因之发展了多种的泛函算法:局域密度近似(LDA)、自旋极化的局域密度近似LSDA、广义梯度近似(GGA)。这三种算法已经获得广泛的应用。基于上述几种算法,目前又发展了精确交换和屏蔽交换、自相互作用修正(SIC)、以及其他交换相关泛函如meta-GGA泛函、PKZB泛函、TPSS泛函、平均密度近似(ADA)及加权密度近似(WDA)等。除了前面提到的这些外,密度泛函理论体系还有许多发展,如常用于材料计算的GW近似、应用于金属氧化物电子强关联体系的LDA+U和动力学平均场理论DMFT、处理任意强度磁场下相互作用电子体系的流密度泛函理论(CDFT)、运用线性响应技术得到密度泛函微扰理论(DFPT)等。
通常所说的DFT,是对基于不含时和非相对论的定态薛定谔方程求解。最近又发展了含时的DFT (TDDFT)和相对论的DFT。TDDFT可以通过线性响应理论求激发态能量,而相对论的DFT可以对某些重元素进行计算。
3.1.2. 半经验法
半经验法(Semi-empirical method)是近似的分子轨道理论计算方法。这种方法是以Hartree-Fock-Roothaan (HFR)方程作为计算体系,以建立在零微分重叠(Zero Differential Overlap, ZDO)近似为基础,借助经验或半经验参数代替分子积分,从而可以大大加快计算速度,当然也会不可避免的牺牲计算结果精度,所以,半经验法不是精确的计算方法。
量子化学计算结果,是以量子化学参数来表征:电荷、轨道能级、轨道电子密度、超离域度、原子极化率、分子极化率、偶极矩、分子极性及能量等的量子化学描述符。量子化学参数,表征分子之间的结合性能和化学反应性能,他们都有明确的物理意义,能够直接基于分子结构进行计算,无需输入其他信息,与实验参数不同的是他们不存在统计学误差,能精确地计算药物分子与受体中心位点的结合能力。
除了分子力学和量子力学计算方法外,近几十年发展了一种基于量子力学和分子力学组合法,称为QM/MM法。我们已经知道,分子力学计算是较为粗糙的计算,但可以计算各种大体系或复杂体系,包括介观(纳米级)和宏观尺度的凝聚态。但分子力学难以对化学反应、过渡态等实质性的问题进行计算。而量子力学基于第一性原理的从头计算法则是精确的计算,是微观尺度上的计算,虽然已经发展了密度泛函理论,但对于介观和宏观体系,仍然无能为力。
3.2. 药物设计中的计算方法
近年来,随着人们对生命科学研究不断深入,需要了解药物分子与人体内受体或酶等蛋白质活性位点的作用机制,以及药物在人体内的转运机制和代谢机制,才能提高药物设计的命中率。同时,一些新材料,特别是某些有特定功能的新材料的研发和拓展应用,需要从分子或原子尺度了解材料的性能,也要从分子或原子的集合体,即凝聚态角度把握材料的性能。虽然现在计算能力不断增强,但是分子模拟在原子水平上对大型复杂系统进行模拟,仍是一个挑战。与此同时,人们对于酶、蛋白质、纳米材料,凝聚相反应和催化系统等的模拟越来越大的兴趣,在较大尺度上模拟系统,来揭示新的材料的细节。
这就涉及到多尺度模拟计算方法问题。近年来,适用于多尺度模拟的计算方法得到了迅速发展。这些计算方法可以以不同的方式来完成多尺度问题,其中之一即是所谓的量子力学和分子力学组合法(QM/MM)。原理上,是将精确的量子力学用低计算成本的分子力学描述,在过去的几十年中已深受欢迎。其基本思想是:用量子力学处理感兴趣的中心,如酶和底物结合的活性位点,其余部分用经典的分子力学方法处理。
这说起来简单,但关键问题是分别应由QM和MM计算时,相互之间有何影响?QM计算和MM计算的边界在哪里?在它们的边界区域采取什么办法计算比较好,才能保证准确、计算速度快、计算费用少?
现在以酶催化的活性位点为例。假设在酶中的活性位点及其附近区域,我们称其为主要的子系统(用PS表示),用QM方法处理,周边环境的影响,例如,蛋白质环境,称为次要子系统(用SS表示),在MM水平上处理。QM/MM的整个系统的能量(ES)被定义为:
即,系统总能量ES为PS的能量,SS的能量,以及它们之间的相互作用能之和。在PS区域和在SS区域上的分子或原子,可以独立进行能量计算,不存在问题。但两个区域的交界处,即PS|SS的计算,就相当麻烦。因为PS与SS区域的电子相互影响,即主子系统(PS)和次子系统(SS)之间存在耦合,这是QM/MM方法的核心问题。处理的PS和SS之间的相互作用的各种各样的QM/MM的方案目前已经得到了发展。
为了解决这个问题,研究人员经过缜密的研究,提出了各种各样的解决方案,如采用机械嵌入法和静电嵌入法,采用在QM和MM计算区域之间增加多个局域,然后通常低层次的QM计算,如半经验的QM计算,或者是采用赝势基组的QM计算。另外,还有采用自适应的QM/MM法,设计原子替换法等等。
总之,计算化学方法是根据计算体系和实际需求,采取不同的计算策略。如:分子力学计算,适应于大体系,获取结构信息。半经验方法,适应于中等体系,获取粗略的性质。从头计算法,适应于小体系,获取准确的性质。密度泛函理论,适应于中等体系,获取准确的性质,QM/MM法可以获得较大体系的较精确计算。
上述介绍的是计算化学的主要方法。除此之外,分子模拟技术也是化学计算的重要组成部分。它是将计算化学从静态性质的描述走向动态性质的描述。
分子动力学模拟(Molecular dynamics simulation),简称为MD模拟,是利用牛顿力学原理来模拟分子体系的运动,以在由分子体系的不同状态构成的系统中抽取样本,从而计算体系的构型积分,并以构型积分的结果为基础进一步计算体系的热力学量和其他宏观性质。分子动力学方法具有以下特征:
① 分子动力学是在原子、分子水平上求解多体问题的重要的计算机模拟,可以预测纳米尺度上的材料中力学特性。
② 通过求解所有粒子的运动方程,分子动力学方法可以用于模拟与原子运动路径相关的基过程。
③ 在分子动力学中,粒子的运动行为是通过经典的牛顿运动方程所描述;
④ 分子动力学方法是确定性方法,一旦初始构型和速度确定了,分子随时间所产生的运动轨迹也就确定了。
分子模拟一般需要三个要素:建立系统模型,建立质点的运动方程和计算分子间的相互势能。
建立系统模型:选择一个由N个粒子组成的分子动力学元胞,加上边界条件构成分子动力学一般模型。研究问题不同,边界条件不同,通常根据具体的研究问题,选取的边界条件有周期性边界条件,固定边界条件,混合边界条件(周期性边界条件加固定边界条件)。
建立质点的运动方程:分子动力学模拟中假设系统所有粒子的运动遵循经典牛顿运动定律,且忽略原子背景电子云的量子效应,原子间的相互作用满足叠加原理。
根据牛顿力学第二定律: ,而作用在第i原子上的总原子力等于其周围所有其它原子对该原子作用力之合力,即: 。
原子间的相互作用力可通过势函数对原子间距离rij求导得出,公式如下:
以牛顿定律为基础,推导出拉格朗日(Lagrangian)运动方程和哈密顿(Hamiltonian)运动方程两个分子动力学运动方程。
计算分子间的相互势能:势函数的研究和物理系统上对物质的描述研究息息相关,不同的物质状态描述用不同的势函数。所谓动力学模型,是由势函数决定的。所以,势能函数的确定,是分子动力学的关键。势能函数与力是随着原子间距的变化而变化。所以其势能是关于原子间距的函数。
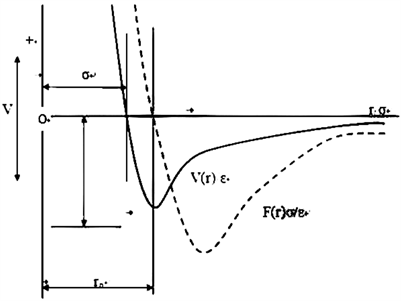
分子动力学模拟的基本过程是在一定系综(如:微正则系综、正则系综、等温等压系综和等温等焓系综,等等)及已知分子势能函数的条件下,从计算分子间作用力着手,求解牛顿运动方程,得到体系中每个分子微观状态随时间的真正变化,再将粒子的位置和动量组成的微观状态对时间平均,即可求出体系的压力、能量、粘度等宏观性质以及组成粒子的空间分布等微观结构。该方法既可计算体系的平衡性质,也可计算体系的各种动力学性质。分子动力学模拟,关键是确定势函数。而势函数基于所研究体系的分子力场。这必然存在严重的缺陷,因为分子力场忽略了电子极化效应。另外分子力场无法描述化学键的形成和断裂的本质问题。因此,一种新的模拟分子方法:从头计算分子动力学方法(AIMD)应运而生,即是最近发展的量子动力学模拟。目前AIMD法主要采用密度泛函理论。由于引入了密度泛函理论,极大地扩展了计算机分子模拟技术的深度和广度,成为计算机分子模拟最先进和最重要的方法之一。
统计力学模拟(Statistical mechanics simulation),简称SM模拟,是一个以最大乱度理论为基础,借由配分函数将有大量组成成分(通常为分子)系统中微观物理状态(例如:动能、位能)与宏观物理量统计规律 (例如:压力、体积、温度、热力学函数、状态方程等)连结起来的科学。如气体分子系统中的压力、体积、温度。伊辛模型中磁性物质系统的总磁矩、相变温度和相变指数。其方法主要是蒙特卡洛方法(即MC法),统计力学根据物质的微观组成和相互作用,研究由大量粒子组成的宏观物体的性质和行为的统计规律。经典力学应用于原子、分子以及宏观物体的微观结构时,其局限性就显示出来,因而发展了量子力学。与之相应,经典统计力学也发展成为以量子力学为基础的量子统计力学。量子统计力学是根据微观世界的量子力学规律改造经典统计力学。量子统计力学的量是相应微观量的统计平均值。根据微观粒子性质和运动力学规律,采用概率统计方法阐明并推断物质的宏观性质和规律性。它包括经典统计力学、量子统计力学、平衡态与非平衡态统计力学等。
总之,计算化学在药物分子设计方面的应用内容十分丰富,包括量子化学计算、分子力学计算、分子动力学计算、分子对接计算及比较分子场分析(Comparative Molecular Field Analysis, CoMFA)和比较相似因子分析(Comparative Molecular Similarity indices Analysis, CoMSA)等 [9] - [27] 。
4. 计算化学的发展
计算化学是一门交叉学科,它和物理化学、分子物理、生物化学、计算科学等多种相关学科都有很强的交叉和渗透 [28] 。其通过应用量子力学和统计力学研究化学问题,与实验性化学不同的是计算化学主要是通过基础理论研究指导化学实验或者验证化学实验结果。理论化学的重要性在于,它研究的是化学学科最核心和普遍的规律,其研究的结果对化学研究中的其他多种学科都有重要的指导意义 [29] 。计算化学的研究对象从小分子和简单理想体系的反应,发展到与其他化学和物理研究领域相关的化学体系、例如生物化学、凝聚态物理学、纳米技术或分子生物学,从而逐步实现对复杂化学体系的理论模拟。随着密度泛函理论的普及,线性标度技术的引入,多参考态方法和量子动力学理论方法的发展,计算化学在我国也有很大发展,许多理论化学家在配位场理论、分子轨道图形理论、价键理论等方面做了大量研究工作并取得了一定的成果。
20世纪80年代是计算化学计算飞速发展的时期。出现了计算化学研究的重要工具Gaussian软件,其是由1998年诺贝尔化学奖得主沃尔特·库恩(Walter Kohn)和约翰·波普尔(John Pople)开发的,它是进行从头算的鼻祖。Gaussian程序是一个最普及的程序,它最早的版本是1970年的Gaussian70,最新的版本是Gaussian09。它可以进行各种类型的从头算,半经验和密度泛函 [30] 。Gaussian程序可以预言分子和化学反应的许多性质,如分子能量和结构,电子密度分布,热力学性质,振动频率,红外和拉曼光谱,NMR化学位移,极化率和静电势等。2013年诺贝尔化学奖得主Martin Karplus (马丁·卡普拉斯)等开发的Charmm软件,Charmm是Chemistry at Harvard Macromolecular Mechanics的缩写,是一个用来进行经典的、量子的或量子经典杂化的模拟计算程序,目前其在科研学术和药物开发商业等领域都得到了广泛应用。在Charmm软件的基础上,Paul Weiner研究组开发了Amber 程序的第一个版本,Wilfred van Gunsteren研究组,开发出了GROMOS程序。NAMD程序,更是基于Charmm进行了并行化的处理,把Charmm的功能发挥的淋淋尽致。此外还有Gamess软件、Amber软件、Vasp软件等都在计算化学领域有重要的应用。例如在模拟药物如何到达体内靶蛋白的实验中,电脑可直接对与药物相互作用的靶蛋白原子执行量子理论计算,精确分析出药物发生作用的全过程。
国内计算化学发展也十分迅速。吉林大学唐敖庆理论化学学派,北京大学唐有琪、徐筱杰、来鲁华实验室和徐光宪、黎乐民实验室,中国科学院上海药物研究所嵇汝运、陈凯先、蒋华良实验室,北京师范大学刘若庄课题组及胡文祥教授课题组等在我国计算化学领域作了许多杰出的研究工作。
本文作者之一的胡文祥,早在二十世纪八十年代中期在中国科学院上海有机化学研究所攻读博士学位期间,就建立了分子力学与量子化学联算的基本思路和方法,博士毕业后到军事医学科学院六所工作,在恽榴红教授支持下与计算机专家合作,编制了自动挑选数据的程序,开创了全新的计算过程,从经典的分子力学计算分子的低能构型构象等几何参量入手,再输入给量子化学程序计算分子的轨道能量和电荷密度等电子参数,或者使用分子力学计算整个分子骨架几何参量,用量子化学计算活性中心的电子参数,如此一来计算效率得到了极大提高。从分子力学计算直接过渡到量子化学计算,发挥了两者的长处,得到了两类结果 [31] [32] 。他应用这一联算方法,研究胆碱能药物空间构象与构效关系等,获得了国家和军队科技进步奖等一系列成果。胡文祥教授实验室继续发扬计算化学方面的优势,进行分子力学与量子化学联算、量子参数与反应性关系、核磁共振化学位移统一计算 [33] [34] [35] [36] [37] 、药物分子二维和三维定量构效关系、分子动力学模拟和分子对接研究等研究工作,不断取得新结果。
5. 计算化学的任务
化学的主要作用之一是为满足人类生存与发展的各种需要而发现或创造制备各种可用性质的化合物。它包含三类:其一是未知化合物结构的测定;其二是具备某种特定性质的化合物分子结构的预测。其三化合物的制备方法。这三类,可以说是化学的三大任务。化学体系是一个具有高度复杂的体系,人们往往在实验的事实基础上,从已知知识(数据)中找出一些共同规律,或从类比推测中来解析这些事实,并形成一系列的理论。而所形成的一系列理论,仅作为解析科学的工具。然而,随着计算机科学的迅猛发展,化学理论和网络技术的发展,为计算化学提供了可行性的发展机会。当前计算机辅助结构解析、分子设计和合成路线设计的研究已经变成十分重要。因为只有通过计算机才有可能对浩如烟海的化学知识进行有效地处理,对结构变化引起的属性变化进行系统地搜索,并用智能程序模仿化学家的思维活动进行高速的推理(分子正确结构的确定、具备某种性质的化合物分子结构的预测和合成路线的确定等)。
计算化学在化学研究领域的应用越来越广泛。典型的应用,包括:计算机辅助分子设计(药物、农药、材料等)、计算机智能化分子结构图谱解析、化学反应及其过渡态理论计算、计算机辅助化工设计、计算机辅助合成路线设计,等等。
5.1. 计算化学任务之一:计算机辅助分子设计
计算机辅助分子设计包含着计算机药物(包括农药等)分子设计和计算机辅助材料设计。
1) 计算机辅助药物分子设计
计算机辅助药物分子设计的目标是先导化合物的发现及其优化方法。其研究方法主要有间接法、直接法两类。
间接法,即基于配体的设计方法。当缺少靶标(受体、酶等生物大分子)结构信息时,主要采用这种方法:从一组小分子化合物(一般需要十至几十个化合物)的结构和生物活性数据出发,研究其结构与活性的关系规律,在此基础上预测新化合物的生物活性,从而进行高活性分子的结构设计。主要是定量构效关系研究(QSAR),包括2D-QSAR (如:Hansch、Free Wilson法)和3DQSAR (如:CoMFA法、CoMSIA法)。这些方法的主要问题在于需要合成一定数量并经过严格的药理学评价获得活性数据的化合物,我们将其称为“训练集”,然后通过机器学习,和统计学计算,获得相关性很好的QSAR方程。然而,我们在研究中经常会碰到一些十分尴尬和困惑的现象,即构建的非常好相关指数的QASR方程,但在预测外部数据集我们称之为“测试集”的活性时,却与实验数据大相径庭。可能存在“活性悬崖”的现象,即微小的结构变动带来活性极大变化,常常因此造成了设计上的失败。这也许是当今计算机辅助药物分子设计的瓶颈。究其原因仍然迫切需要人们探究和解析。
直接法,即基于受体的设计方法。已经获得靶标(生物大分子,如蛋白质、核酸、酶、离子通道等)的结构信息,来研究药物分子与受体的相互作用,从而获得药物分子的结构特征,设计出从空间形状和化学性质两方面都能很好地与靶标分子相结合的药物分子。随着分子生物学、细胞生物学和结构生物学的发展,越来越多的药物作用靶标分子(蛋白质、核酸、酶、离子通道) 被分离、鉴定,其三维结构被阐明,为直接药物设计方法的应用提供了有利的条件。直接药物设计包括全新药物设计(de novo drug design)和数据库搜寻(或称分子对接,Docking)两类。全新药物设计,是根据靶标分子与药物分子相结合的活性部位的几何形状和化学特征,设计出与其相匹配的具有新颖结构的药物分子。数据库搜寻,首先应具备大量化合物(例如几十至100万个化合物) 的三维结构数据库基础上,将库中的分子逐一与靶标分子进行“对接”(Docking),通过不断优化小分子化合物的位置(空间取向)以及分子内部柔性键的二面角,寻找小分子化合物与靶标大分子作用的最佳构象,计算其相互作用及结合能。在库中所有分子均完成了对接计算之后,即可从中找出与靶标分子结合的最佳分子。
目前的药物设计主要是一种基于药物和靶标生物大分子三维结构的设计方法。即人们想知道的是药物分子是怎样与生物大分子结合的,它们的结合对人体会产生怎样的生理效应。然而,药物在人体的作用过程是非常复杂的系统。一个优良的药物除了与靶标分子产生所预期的相互作用之外,还应该具有良好的体内输运和分布性质以及良好的代谢性质,而这些要求在基于结构的药物设计方法中未能予以考虑,而且,也未考虑药物分子与受体活性中心结合是否对受体其它部位的结构变化而产生活性变化。随着新世纪生命科学、计算机科学的发展,考虑药物作用不同机理和全部过程的药物设计—基于作用机理的药物设计方法(Mechanism—Based Drug Design)将逐步建立和完善。未来的药物设计,可能将从基于结构的药物设计向基于作用机理的药物设计方向发展。基于作用机理的药物设计,离不开计算化学,特别是量子力学,同时还需要化学、物理、生物等学科之间的紧密结合。虽然目前基于机理的药物设计还处于起步阶段,尚未形成系统的理论体系。
不管是间接法还是直接法,都要进行配体与受体的相互作用计算。它包含了上述所及的量子化学计算和分子模拟计算的各种方法。目前,被认为最为重要的方法是量子力学和分子力学(QM/MM)相结合的方法:药物分子与底物分子(酶、蛋白质分子)的结合中心位点,采用精确计算的量子化学QM法,而外围部分采用经典的分子力学MM的高效方法。这种结合的方法,目前正在发展中,而且已经取得一定的成果。
我们相信,随着新技术的不断涌现,药物在体内的作用形式、作用机理和性能的理论研究不断进步,新的统计学方法的引入,如支持向量机(SVM)新的机器学习方法的应用(小样本、非线性及高维模式识别),不久将来,计算机辅助药物分子设计的“瓶颈”将会突破,那时,计算机药物设计的辉煌时代将到来。
2) 计算机辅助材料设计
目前计算化学发展最快、最为活跃、应用面最广的可以说是计算机辅助材料设计,而且已经取得了许多丰硕的成果。其主要包括特殊材料功能的设计、材料内部结构和特征的模拟与优化、材料内部损伤和晶体缺陷等对材料性能的影响计算和模拟仿真,等等。
目前材料研究主要集中在凝聚态,并且尤以固体材料研究为重点。固体材料从结构上大致分为两类:致密固体材料和多孔固体材料。致密固体材料,主要是磁,电,多铁性等性能的材料,包括热电材料,超导材料,铁电材料(多铁性材料),储氢材料(能源材料)、复合材料(纳米隐身复合材料)等。多孔固体材料,主要是能量转换与储存性能的材料,如:催化剂、蓄电池材料、储氢材料、燃料电池、光电池、发光材料、透明导体、隐身材料等。
材料的特性与原子或分子的结构、排列及其运动状态密切相关。因此,要研究材料的特性,必须掌握材料组成、内部微观粒子(原子)的空间架构和运动规律,理解它们对材料的变形与破坏的机理。但由于原子尺度在埃量级,直接观察非常困难。在这种背景下,对材料的原子尺度进行数值模拟就显得越来越重要。
利用计算机技术进行材料设计是发展新型材料的重要手段。材料设计通常分为3个层次。第一个是微观层次,即运用量子力学与量子统计力学来研究原子或分子的集体行为。第二个是介微层次,其大小在微米以上,研究的是许多原子或分子在一定范围内的平均性质,如形变、磁性等,一般用连续统计方程来描述。第三个层次是宏观层次,如宏观性能、生产流程与使用性能间的关系,材料的断裂以及微观结构的形成等一般采用分子动力学模拟。计算机技术可以把3个层次的因素都考虑在内,通过建立模型(获取不同层数的数值参数,需要分别建立与之相适应的计算模型),然后进行计算和分子模拟。在此基础上,通过进行原子(或分子)替换和结构优化,得出符合预期性能的新材料的最佳成分、最佳结构以及最合理的工艺流程。计算机的高速计算能力、巨大的存储能力和逻辑判断能力与人的创造能力相结合,可对材料设计提出创造性的构思方案。
在材料研究方面,越来越多地采用QM/MM法。QM方法可以应用于研究化学反应,配位数的变化,而MM的方法可应用于经典粒子间的相互作用。在使用QM/MM方法时,重要的是,需要仔细考虑QM方法结合在MM的环境。
研究晶体(包括导体、绝缘体和半导体的晶体)电子的状态及其运动性质,离不开能带理论。能带理论认为晶体中的电子是在整个晶体内运动的共有化电子,并且共有化电子是在晶体周期性的势场中运动;结果得到:共有化电子的本征态波函数是Bloch函数形式,能量是由准连续能级构成的许多能带。能带理论在阐明电子在晶格中的运动规律、固体的导电机构、合金的某些性质和金属的结合能等方面取得了重大成就。为了更准确的能带结构的方法的需要,必须开发更有效的算法。
3) 构效关系研究
世界万物的性质在一定条件下是由其结构决定的,这就是构效关系研究的基础。无论是药物还是材料,有什么样的结构,就可能具有什么样的性质和效果。药物的构效关系分析始于20世纪60年代Hansch所建立的方程
后来发展了分子力学和量子化学方法计算的参数与药物的药效进行相关分析,建立了含有量子参数的构效关系方程,使这方的研究又前进了一大步。20世纪80年代末90年代初,又推出了比较分子场分析等三维定量构效关系方法,是构效关系研究又上一个新台阶。
目前药物分子设计与构效关系研究,正在结合组合化学、高通量与高容量筛选及机器人筛选等新技术,推动有机化学及药物化学日新月异的发展。
5.2. 计算化学任务之二:计算机辅助智能化化学结构解析
化学结构解析,即未知化合物结构的测定。它包含两个方面的内容。其一是合成化合物的结构测定。由于合成化合物是人们按照可能存在某种符合自己设想性能的化合物结构进行设计,所设计的化合物一般都是根据已有的化学反应知识和合成规则进行合成。由于有前体化合物的结构已知,最终化合物的结构分析可以通过几种常规的波谱分析表征,这是比较简单的结构解析。其二是已知其性能的某种化学物质,如某些天然产物、蛋白质、核酸等,只知其性能而不知其化学结构和组成。他们通常是通过提取分离、或生物制备获得的产物,往往结构十分复杂,解析其化学结构十分困难,需要多种现代分析仪器进行结构测定。现代分析仪器发展已经十分成熟的今天,即使是经验十分丰富的化学家,有时对复杂的未知化合物的结构图谱解析也束手无策。
计算机辅助解析化学结构,已经引起了人们的高度重视。它包括两个方面的内容。其一是在某一或联合使用的分析仪器,加入计算机辅助分析软件,来帮助人们识别谱图,获得化学结构式。其二是独立的计算机结构分析系统,它是将各种分析仪器获得的光谱数据进行智能化谱图解析,自动获得化合物结构。
5.3. 计算化学任务之三:计算机辅助合成路线设计
传统的化合物合成设计通常是化学工作者根据丰富的经验和灵感来完成工作。但是今天,以逻辑的方法而不是单凭经验和直觉来设计合成路线已经成为化学家设计合成路线的一个重要手段。虽然目前计算机合成路线设计还是初步的、辅助的,但已经迈出了这一步,经过几十年的发展和完善,可以预见,计算机辅助合成路线设计将变成最重要的手段,到时可以将“辅助”两个字去掉,成为化学合成路线设计的主要手段。
计算机辅助合成设计,包含了两个基本方式:其一是检索型的合成设计。即在现有的化学反应数据库中进行检索。尽管已有大量数据可供参考,化学反应体系的高度复杂性决定了难于用纯理论方法来解决合成路线设计问题,还只能从已知知识中找出共同规律,或从类比推测中来近似地解决这些问题。其二是推理型的合成设计。包含反应数据挖掘和反应知识发现、反应知识模型的表述和反应知识库的建立、化合物反应性能的预测、化学知识的类比推理等。
检索型合成设计,是基于现有的化学反应数据库。随着计算机科学和技术的发展,计算机在化学领域中得到越来越广泛的应用。从最初的简单数据处理,逐步发展到对化学结构的处理,以及目前的对化学信息的综合分析、知识获取和应用。其中化学数据库对化学工作者有着非常重要的作用和意义。化学反应数据库是从事化学合成的科技人员获取化合物合成信息的有效途径。数据库中存储着大量的已合成或发表的化合物的合成信息,根据这些信息,计算机通过来选择设计新的合成路线。同时人们凭经验和知觉,或者通过计算机分析来优化合成路线,达到合成步骤少、合成总产率高、原料简单便宜易得的目的。
推理型的合成设计,是基于计算机辅助合成设计系统,它是由反应知识库和反应合成分析两部分组成。首先计算机对用户提供的化合物结构进行分析,以确定该化合物的合成反应策略;再利用反应知识库中的反应知识和反应规则分析,以确定该化合物的前体。循环进行这二步操作,以最终获得起始反应原料为止。通过对这个循环过程路线的记录,便可得到一组可参考的该化合物合成路线的信息,这些信息辅助化学工作者的合成设计工作,以提高他们工作效率,并降低各个方面的消耗。
反应知识库,是一个开放式的数据库,在拥有目前已知的化学反应知识,包括的化学反应类型、反应机理、合成方法等,同时对于新发现的反应知识不断扩充到库中。而反应合成分析,是计算机对人们输入计算机的化合物结构,经过合成反应策略分析和确定后,通过反应知识库分析和所建立的数学模型的求解演算进行智能化的取舍,经过记录、分析、推断、归纳、判断这一整体的分析系统,即是逻辑推理过程,最后获得若干个反应路线。人们可以根据自己的合成实践经验、方法的熟练程度、实验室或工业生产的合成条件、原料性质和原料的价格,通过人机对话,优选一条合成路线。
未来的计算机合成路线的发展,除了进一步对推理型的合成设计充实和完善外,还应当对包含化学反应动力学的分析,即对所设计的合成路线的每一步反应,提出最佳的反应条件,如:反应物的摩尔比、催化剂选择和用量、溶剂选择和用量、反应温度、反应压力、反应时间、隔氧隔湿建议、可能的副产物、产物提取方案和纯化方案等等,甚至还要提出注意事项特别是反应过程容易发生易燃易爆有毒等危险的注意事项。
6. 计算化学应用前景
通过对具体的分子系统进行理论分析和计算,能比较准确地回答有关稳定性、反应机理等化学问题。目前计算化学已被广泛用于化学合成、材料、催化和生物化学等研究领域。
计算化学在生物化学方面的应用,有金海晓等 [38] 采用计算化学方法探明蛋白激酶在水溶液中的动态行为,包括磷酸化对蛋白激酶构象的影响、磷酰基转移机理以及蛋白激酶与抑制剂的作用模式等,这些工作为合理设计药物提供了参考。此外,曹冉等 [39] 对计算化学方法在基于受体结构的药物分子设计中的多种应用,包括小分子结合位点的成药性评估、化合物数据库的虚拟筛选、先导化合物的结构优化等进行了探讨。
计算化学在化学合成上多用于化合物的理论计算结果与其实验结果的对比。如邢波等 [40] 用GaussView软件模拟苯的分子结构,用量子化学计算软件Gaussian 03W的密度泛函法,在B3LYP/6-31G基组水平上,优化苯分子结构、计算能量和频率,及其红外光谱,并与实验结果对比。根据成键轨道(natural bond orbital, BO)和电子密度计算结果,探讨苯的稳定性,分子轨道能量,原子静电荷分布规律和前沿分子轨道组成的特征,计算得出HOMO和LUMO的能量差是0.25191 eV,HOMO是比较大的负值(−0.24819 eV),说明苯有较好的稳定性,看来采用纳米TiO2光催化方法降解苯是可行的。邢波等还 [41] 通过应用计算化学软件Gaussian 03WHF方法中的3—21G基组优化苯和甲苯分子结构,预测苯和甲苯分子的红外光谱。找到苯环振动吸收峰分别是苯红外图中的1658 cm。与甲苯红外图中的1667 cm~。与苯和甲苯文献检索红外谱图相对应(特征吸收峰分别是1478和1485 cm−1),符合较好。还找到苯环C-H拉伸振动吸收峰3080 cm−1 (苯)和3040 cm−1 (甲苯)。此外,还有对多种类似化合物进行化学计算进行对比,如盛旭玲 [42] 采用Materials Studio软件中的DMol^3模块,对18种苯的二取代在药物研发过程中手性化合物绝对构型的确定是一个极其重要的问题,药物分子的立体构型与药物的疗效、毒性密切相关,因此,药物分子立体结构的研究具有重要意义。目前,手性化合物立体结构的研究方法也倍受关注,运用计算化学方法确定手性化合物的立体构型是一种简便而有效的方法,已经被越来越多的研究者接受并采用。王重娟等 [43] 利用计算化学方法,对旋光计算、电子圆二色谱(ECD)计算、振动圆二色谱(VCD)计算,以及13C-核磁共振(13C-NMR)计算等方法进行综述。为了在化学领域中更好地应用微波能,需要深入研究微波与化学反应相互作用的机理,由于等效介电系数常常被用来描述在化学反应中微波的吸收和传输情况,因而,化学反应中反应物的等效介电系数就成为微波化学研究的关键问题之一。华伟等 [44] 基于反应溶液中单位体积内的分子个数随温度变化的情况对黄卡玛提出的经验公式进行了改进。以碘化钾和高锰酸钾两个氧化还原反应为例,对实验结果和计算结果进行了分析比较,结果表明其具有很好的一致性,而且计算精度较改进前的公式得到了改善。
计算化学在对天然产物绝对构型的确定上也有重要应用。由于传统方法在天然产物绝对构型确定方面存在一定局限性和不足,而大部分工作交由计算机完成的计算化学方法,在保持同样高精确度的基础上,可以节约大量时间。李桢等 [45] 文章中有多个应用两种或多种计算光谱法相结合在天然产物绝对构型确证的实例,表明计算化学方法在天然产物研究中已经逐渐被认可。
目前计算化学的研究已经取得了很大的成果,其对化学学科的发展影响是巨大的。随着化学理论的不断发展,以及计算机性能的迅猛发展,计算化学通过对涵盖若干公理的一个系统方程的求解,解决化学的问题。它不依赖传统化学实验仪器设备、试剂和药品,是建立在理论的演绎思维的基础上的电脑模拟。计算化学的第一原理是具有公理结构的,经过数学和逻辑演绎而得到关于物质的形式理论体系,再从形式理论出发利用物理假设出发,利用物理模型近似,二次形式化和计算,得到理论预计值,最后在再去与实验结果核对。因此,以量子力学,统计力学为核心的计算化学在理论化学与实验化学之间起到了很大作用。
7. 虚拟现实
计算化学是理论化学的执行者,而根据现实的化学实验环境编制的仿真性软件在计算机中营造一个接近真实环境的虚拟场景来完成各种预定的实验任务,可以验证理论模型,同时可以帮助科研人员理解、预测和发现新的化学现象和物理本质。这就是计算机虚拟化学实验室。这里包含着量子化学计算和分子模拟技术,对化学和生命科学领域的某些未知或未定问题进行科学模拟,它是在在特定数值模型的基础之上,对现实中的化学反应过程、物理化学过程、生命过程、化学工程的生产流程等实现仿真模拟。
各种材料都有其特性在,掌握原子结构、排列及其运动规律,对理解材料的变形与破坏的机理是至关重要。但由于原子尺度原因,人们目前尚无法直接进行观察。因此,对材料的多尺度进行数值模拟和仿真模拟都十分必要。
一种叫做“触觉量子化学”,它是通过一个量子力学的计算模型程序结合到计算机感应装置来反馈用户的触觉动作的虚拟仿真。例如,你手中握着一个笔式的触觉设备。当你把一个分子移向另一个分子的时候,该设备将会把你的移动反馈给计算机,此时计算机立即计算出分子之间的吸引力或排斥力。这有点类似于人们在网络上打台球时用鼠标给台球施加力(长按给力健)台球的运动速度就加快感觉。于是,人们具有能够操纵系统原子核坐标的势能面的曲率的触觉感,让你以一个非常直观的方式觉察到分子的复杂信息:分子结构、原子能量梯度、原子轨道、电子密度等等其它性质。这种力的计算也将会让你感觉到一个优先的反应路径应该是什么,那么你就会考虑应该怎样做。这显然是一个令人兴奋的设想。因为,触觉量子化学方法能够把人们引进了更深层次科学问题的理解。目前已经有一些这样可用的方法,但他们只用经典力场来计算由设备表现的力,无法实现化学反应过程的模拟。由于化学反应存在原子键的形成和断裂问题,只有采用精确的量子力学计算才能实现这一过程。然而量子力学计算量大,十分耗时,以目前的技术水平难以实现真正的“触觉量子化学”。于是,一种新的量子化学计算方法浮出水面,叫做“实时量子化学”。
为了实现对研究对象进行既快速又高精确性的计算,“实时量子化学”应运而生。其是通过鼠标、键盘或专用输入工具来驱动所研究体系中分子结构的操作,计算机将根据量子力学的波函数和相应能量及其梯度的活性分子体系的快速计算,其计算结果获得实时响应,操作者感觉到没有延迟的数据流。“实时量子化学”实现实时响应,需要两个方面条件:其一是硬件条件,需要多核并联的中央处理器(CPU)和专用的图形处理器(GPU),来保证显示刷新间隔在100毫秒之内的速度;其二是波函数求解算法的优化,尤其是在密度泛函理论框架内自洽场(SCF)迭代快速收敛方法,同时需要选择合适的基组来加快计算速度的办法。最理想的是量子计算机的应用,可以真正满足“实时量子化学”的快速计算要求,从而实现“触觉量子化学”的化学反应过程的实时模拟和动态仿真。实时量子化学,可能改变未来如何进行量子化学的研究方式。触觉量子化学,是人类更直观地对现有的科学数据的触觉感开发迈开的第一步,通过应用技术的更深嵌入,已经在虚拟现实领域朝发展这个终极目标方向进发。
8. 展望
在过去的50年里,计算化学的发展如火如荼,已将化学带入一个新时代,计算化学促进了现代化学研究领域的一场革命性的变化。当前,计算化学已经深入到化学研究领域的方方面面,正在或者将要起到“领军”作用。下一步,计算化学将面临更复杂更新的挑战。
目前薛定谔方程的求解,都是基于电子结构理论并应用各种简化算法方案进行求解。而含时的薛定谔方程的求解、相对论的薛定谔方程求解等工作正在展开,并开始在各方面应用。在过去的二十年中,量子化学的另一个分支:基于原子核运动理论的薛定谔方程,已经获得不含时的变分法解决方案,并快速的发展,多原子体系的核运动的薛定谔方程如同电子运动的势能面(PES)一样的“精确”。它宣告,我们现在已经处在量子化学的第四阶段。第一到第三阶段主要是以电子结构技术为主体的发展阶段,而第四个阶段,则是以原子核运动为主体的发展阶段。在这个阶段中,我们以一个准确的方式将其纳入到量子化学来处理原子核运动。基于电子运动和基于原子核运动的量子化学联合求解方法,将会获得更多的分子结构和分子运动信息,也必将在各个应用领域获得突破性发展。
量子计算机的诞生,必将大大缩短了计算成本,从而使从头计算的范围大幅度的增加:从几百个原子的微观体系扩大到上万个原子聚合体计算,宏观体系的量子化学计算将成为可能。这意味着对蛋白质、核酸等大分子的生命载体和遗传信息载体的空间结构研究,将会有突破性进展。
随着后基因组时代的到来,人们开始把研究的重点转向了功能基因组,即从基因、蛋白质中研究生命的本质,理解结构与功能、发育与疾病的关系。随着计算机技术速度的大幅度提高,特别是量子计算机将要问世,“从头预测(Ab initio)”方法将可能成为蛋白质空间结构的主要预测方法。因为它不依赖于已有的结构数据信息,直接从蛋白质序列利用分子动力学原理预测和推断结构信息。从而可以揭示蛋白质的结构与功能的关系、总结蛋白质结构的构成规律、预测蛋白质肽链折叠和蛋白质的结构等。同时,从头预测方法与蛋白质的比较建模(Comparafire mode)、折叠识别(Fold recognition)、网络模型和基于隐马尔可夫模型的机器学习方法等同源模建方法相比,延伸到了生命现象的核心。从蛋白质序列和三维结构中预测其功能信息,是后基因时代的一个重要课题。但目前尚需要解决的是:蛋白-蛋白相互作用;肽和肽模拟物;分子多样性和药理空间;分子药效学(意义、潜力和挑战);早期阶段的临床疗效和安全性等。
21世纪的化学不仅要面对简单体系,还要面对包括生命体系在内的复杂系统。因此,除了研究分子的成键和断键,即研究离子键和共价键那样的强作用力之外,还必须考虑复杂体系中的弱相互作用力,如氢键、范德华力等等,而目前的密度泛函理论尚不能解决这一问题,需要进一步发展。虽然氢键、范德华力等作用力较弱,但由此却组装成分子聚集体和分子互补体系。这种超分子体系常常具有全新的性能,或者可使通常无法进行的反应得以进行。基于分子识别观点进行设计、合成及组建新的、有各种功能的分子、超分子及纳米材料,将是未来一段时间中化学的重要研究内容。而深入研究控制分子的各种作用力,研究它们的本质并深刻了解分子识别,是一个颇具重大意义也是一个充满挑战的课题。
生物大分子体系的量子化学计算一直是一个具有挑战性的研究领域,尤其对生物大分子体系的理论研究具有重要意义。由于量子化学可以在分子、原子、电子水平上对体系进行精细的理论研究,是其它理论研究方法所难以替代的。因此要深入理解有关酶的催化作用、基因的复制与突变、药物与受体之间的识别与结合过程及作用方式等,都很有必要运用量子化学的方法对这些生物大分子体系进行研究。毫无疑问,这种研究可以帮助人们有目的地调控酶的催化作用,甚至可以有目的地修饰酶的结构、设计并合成人工酶;可以揭示遗传与变异的奥秘,进而调控基因的复制与突变,使之造福于人类;可以根据药物与受体的结合过程和作用特点设计高效低毒的新药等等,可见运用量子化学的手段来研究生命现象是十分有意义的。
化学键究竟是如何断裂和重组的? 分子是怎样吸收能量的?并是怎样在分子内激发化学键达到特定的反应状态的?复杂体系的化学动力学、非稳态粒子的动力学、超快的物化过程的实时探测和调控以及极端条件下的物理化学过程都已经成为重要的研究方向。向生命学习,研究生命过程中的各种化学反应和调控机制,正成为探索反应控制的重要途径,真正在分子水平上揭示化学反应的实质及规律将指日可待。
理论化学与功能基因组学的结合:将为预测和在分子水平上理解蛋白质的功能,奠定了坚实的基础。利用在蛋白质结构的特殊残基中的固有的,特殊的,化学的和静电的性质,进行催化或识别。这些不同寻常的性质可以通过计算获得,因为蛋白质的活性位点和结合位点可以通过三维结构精确预测。
目前,计算机的飞速发展,使计算化学在各行各业得到广泛的应用,逐渐与各相关学科形成了一些崭新的边缘或交叉学科,如微观反应动力学,量子催化、量子电化学和量子生物学等。与生命科学、材料科学的结合将会更加有力的推动分子生物学、药物设计、新材料的“分子设计”向纵深发展。同时,随着学科之间的交叉和相互渗透,也将启发我们参考、借鉴其它学科的研究方法,使其相互结合,扬长避短,走向一个新的高度。
总而言之,计算化学的目的在于理解、预言和发现新的化学现象及其物理本质。计算化学已经、正在引领未来的化学发展。因为,量子属于物理的、也属于化学的,属于量子世界的,也属于经典世界的。
文章引用
秦 宁,闵 清,李 博,马密霞,邵开元,胡文祥. 计算化学相关研究进展
Progress in Computational Chemistry[J]. 交叉科学快报, 2018, 02(04): 111-131. https://doi.org/10.12677/ISL.2018.24021
参考文献
- 1. Truhlar, D.G. and Mckoy, V. (2011) Computational Chemistry. Springer, Netherlands.
- 2. Wang, L., Hu, W.X. and Liu, X.L. (2009) The Modeling of Three-Dimensional Structure of Human μ-Opioid Receptor and the Study of Molec-ular Docking of Fentanyl Analogs. Computers and Applied Chemistry, 26, 746-750.
- 3. 苏培峰, 谭凯, 吴安安, 等. 理论与计算化学研究进展[J]. 厦门大学学报(自然版), 2011, 50(2): 311-318.
- 4. 任洁, 朱华结. 计算化学在手性化合物结构分析中的应用[J]. 高等学校化学学报, 2009, 30(10): 1907-1918.
- 5. 赵丽娇, 钟儒刚, 甄岩. 计算化学软件在大学有机化学教学中的应用[J]. 计算机与应用化学, 2008, 25(8): 1035-1037.
- 6. Clementi, E., Corongiu, G., 帅志刚, 等. 从原子到大分子体系的计算机模拟——计算化学50年[J]. 化学进展, 2011, 23(9): 1795-1830.
- 7. 郑文锐. 计算化学在化学化工中的应用(上) [J]. 上海化工, 2009, 34(12): 22-24.
- 8. 郑文锐. 计算化学在化学化工中的应用(下) [J]. 上海化工, 2010, 34(1): 23-26.
- 9. Zhang, Z.Y., An, L.Y., Hu, W.X. and Xiang, Y.H. (2007) 3D-QSAR Study of Hallucinogenic Phenylalkylamines by Using CoMFA Approach. Journal of Computer-Aided Molecular Design, 21, 145-153. https://doi.org/10.1007/s10822-006-9090-y
- 10. Zhu, H.W., Fang, H., Wang, L.Y., Hu, W.X. and Xu, W.F. (2008) 3D-QSAR Study with Pharmacophore-Based Molecular Alignment of Hydroxamic Acid-Related Phosphinates that Are Amino-Peptidase N Inhibitors. Drug Discoveries & Therapeutics, 2, 192-197.
- 11. Liu, M. and Hu, W.X. (2009) Research on Structure-Activity Relationships of Fentanyl Analoges with Human μ-Opioid Receptor. The 13th Asian Chemical Congress, Session: Chemical Biology and Medicinal Chemistry, Shanghai, 14-16 September 2009.
- 12. 刘小利, 王陆瑶, 胡文祥. μ阿片受体的三维结构预测及活性位点分析[J]. 化学通报, 2009, 72(2): 133-137.
- 13. Hu, W.X. and Liu, X.L. (2009) Molecular Docking and 3D-QSAR Studies of 4-Phenyl-Piperidine Derivatives as μ Opioid Agonists. The 13th Asian Chemical Congress, Session: Chemical Biology and Medicinal Chemistry, Shanghai, 14-16 September 2009.
- 14. 李博, 刘明,胡文祥. 芬太尼类化合物与阿片μ受体相互作用的分子对接与分子动力学模拟[J]. 物理化学学报, 2010, 26(1): 206-214.
- 15. Liu, M., Wu, Q.S. and Hu, W.X. (2011) Pharmacophore Screening on Piperidinecarboxamides Derivatives Based on GALAHAD and CoMFA Models. Chinese Journal of Chemistry, 29, 1075-1083. https://doi.org/10.1002/cjoc.201190204
- 16. Liu, M., Liu, J. and Hu, W. (2011) Molecular Docking of G. pen-taphyllum to LXRs Receptors and Agonist Screening. The 2nd International Conference on Artificial Intelligence, Management Science and Electronic Commerce, 8-10 August 2011, Vol. 8, 6684-6687.
- 17. Liu, M., Sun, Z. and Hu, W. (2012) Three-Dimensional Pharmacophore Screening for Fentanyl Derivatives. Neural Regeneration Research, 7, 61-65.
- 18. Liu, M., Liu, X. and Hu, W. (2012) Molecular Docking and 3D-QSAR Studies of 4-Phenylpiperidine De-rivatives as μ-Opioid Agonists. Advanced Material Research, 361-363, 263-267.
- 19. Liu, M. and Hu, W. (2012) De-sign, Synthesis and Biological Evaluation of Novel Fentanyl Analogues as Agonist of μ-Opioid Receptor. Applied Mechanics and Materials, 217-219, 937-940. https://doi.org/10.4028/www.scientific.net/AMM.217-219.937
- 20. Liu, M., Wang, L., Liu, X.L. and Hu, W.X. (2013) Study of Molecular Docking of μ Opioid Receptor Agonist Fentanyl and Its Analogs Based on Docking. Ad-vanced Materials Research, 655-657, 1931-1934. https://doi.org/10.4028/www.scientific.net/AMR.655-657.1931
- 21. Lu, J.X., Lei, L., Huan, Y., Li, Y.Q., Zhang, L.J., Shen, Z.F., Hu, W.X. and Feng, Z.Q. (2014) Design, Synthesis, and Activity Evaluation of GK/PPAR γ Du-al-Daret-Directed Ligands as Hypoglycemic Agents. ChemMedChem, 9, 922-927. https://doi.org/10.1002/cmdc.201400009
- 22. 胡文祥, 刘明. 阿片受体分子药学[M]. 北京: 化学工业出版社, 2014.
- 23. 何华军, 王刚, 刘亚军, 谢艳蓉, 沈喜洲, 邵开元, 胡文祥. 苯二氮卓类催眠药物三维定量构效关系研究[J]. 药物化学, 2016, 4(4): 25-37. https://doi.org/10.12677/HJMCe.2016.44004
- 24. Shen, X., He, H., Yang, B., Zhao, Z., Shao, K. and Hu, W. (2017) Studies on the Activities of Electrophilic Sites on Benzene Ring of 4-Substituted Anilines and Their Acyl Compounds with Multiphilicity Descriptor. Chemical Research in Chinese Uni-versity, 33, 773-778. https://doi.org/10.1007/s40242-017-7112-z
- 25. 邵开元, 何华军, 王刚, 刘亚军, 沈喜洲, 胡文祥. 化学势变化率对催眠药类化合物QSAR影响[J]. 化学通报, 2017, 80(11): 1061-1066.
- 26. Liu, M. and Hu, W. (2017) Using Deep Belief Network and Computation Methods to Improve Opioid Receptor Biological Activity Prediction, Novel Agonists and Antagonists Structural Modeling. In: Hu, W.X., Ed., Catalytic Synthesis and Substituent Effect, Hans Publishers, Wuhan, 325-330.
- 27. 刘明, 马密霞, 韩谢. 书评: 《比较化学——构筑量子化学通向分子药学的桥梁》[J]. 比较化学, 2018, 2(1): 6-10. https://doi.org/10.12677/CC.2018.21002
- 28. 殷伟. 计算机在化学化工中的应用[J]. 中国科技信息, 2005(2): 26.
- 29. 杨祖杰. 计算流体力学在化学工程中的应用[J]. 化工设计, 2018, 28(5): 3-5.
- 30. Han, X., Shao, K. and Hu, W. (2018) Synthesis of 9-Substituted Berberine Derivatives with Microwave Irradiation. Chemical Research in Chinese University, 34, 571-577.
- 31. 焦克芳, 胡文祥, 鲁中正, 恽榴红. 分子力学MMPM与量子化学MNDO联算[J]. 军事医学科学院院刊, 1992, 16(2): 159.
- 32. 胡文祥, 恽榴红, 司伊康, 黄量. 棉酚及其相关化合物的分子力学和分子图形学研究[J]. 中国药物化学杂志, 1992, 2(2): 28-30.
- 33. 胡文祥, 恽榴红, 阮金秀. 有机磷化合物核磁共振屏蔽结构效应研究进展[J]. 军事医学科学院院刊, 1993, 17(3): 221-224.
- 34. 胡文祥. 有机磷化合物31P NMR及其规律I.31P NMR化学位移新的经验规律和原理[J]. 军事医学科学院院刊, 1993, 17(1): 1-6.
- 35. 胡文祥. 有机磷化合物31P NMR及其规律II.几类开环有机磷酸酯31P NMR化学位移的统一计算[J]. 军事医学科学院院刊, 1993, 17(2): 93-97.
- 36. 胡文祥. 非环亚磷酸酯与双环亚磷酸酯亲核活性及化学位移的比较研究[J]. 军事医学科学院院刊, 1993, 17(2): 158-159.
- 37. 胡文祥, 袁承业. 双环磷酸酯类化合物NMR结构效应研究[J]. 波谱学杂志, 1993, 10(4): 361-369.
- 38. 金海晓, 严小军, 朱鹏. PKA酶及其抑制剂Balanol的计算化学[J]. 化学进展, 2010(5): 993-1001.
- 39. 曹冉, 李伟, 孙汉资, 等. 计算化学方法在基于受体结构的药物分子设计中的基础理论及应用[J]. 药学学报, 2013(7): 1041-1052.
- 40. 邢波, 刘艳菊, 张婷婷, 等. 苯的计算化学红外光谱表征及其与实验结果对比[J]. 计算机与应用化学, 2010, 27(4): 461-464.
- 41. 邢波, 孙珍全. 采用计算化学方法解析苯和甲苯红外谱图[J]. 现代仪器与医疗, 2010, 16(1): 42-44.
- 42. 盛旭玲. 二取代苯环上亲电取代定位的计算化学模拟[J]. 大学化学, 2011, 26(3): 44-46.
- 43. 王重娟, 王立波. 利用计算化学方法确定手性化合物立体构型研究进展[J]. 中国药物化学杂志, 2013(3): 235-242.
- 44. 华伟, 杨晓庆. 计算化学反应中等效介电系数经验公式的改进[J]. 化工学报, 2006, 57(9): 2111-2115.
- 45. 李桢, 杨洁, 马阳, 等. 计算化学在确定天然产物绝对构型中的应用[J]. 国际药学研究杂志, 2015, 42(6): 751-761.
NOTES
*通讯作者。