Hans Journal of Computational Biology
Vol.
09
No.
03
(
2019
), Article ID:
32377
,
22
pages
10.12677/HJCB.2019.93009
Two-Dimensional Visualization Analysis of Homo Sapiens Gene Sequences
Huaxian Zheng, Zhijie Zheng*, Liuyun Du, Zhongwei Zhang
School of Software, Yunnan University, Kunming Yunnan

Received: Sep. 6th, 2019; accepted: Sep. 20th, 2019; published: Sep. 27th, 2019

ABSTRACT
Chromosomes are the carriers of genetic information. The number of somatic chromosomes in normal human is 23 pairs, and they have certain morphology and structure. Human beings have lost tens of thousands of genes in the long-term evolution process from apes to humans. Studies have shown that the missing parts of genes can reflect human selection in the evolutionary history. Scientists believe that the loss and replication of DNA may be an important evolutionary motive force. It is of great significance to study these gene changes. The number of Y chromosome deletions in humans is obvious. Millions of years ago, there were about 1500 genes on Y chromosome, but now there are only 40. Gene deletion is a kind of gene mutation. Gene mutation can occur at any stage of development. There are many diseases caused by gene mutation in human. Gene controls the expression of biological traits. Gene sequence from expression to protein is related to the sequence of RNA, ncRNA, cDNA, CDS. This paper is devoted to exploring the relationship between these sequences and visualizing these gene sequences in a visual form. In this paper, the variable probability statistical graphical representation method is used to measure the fragmentation probability of the special sequences of chromosome 2, Y, mRNA, CDS, cDNA and ncRNA in Homo sapiens and map them into several 2D probability statistical maps, so as to compare and analyze the distribution of different gene sequences.
Keywords:Chromosome Sequence, mRNA, ncRNA, cDNA, CDS, Visualization, Probability Measurement
人类基因序列二维可视化分析
郑华仙,郑智捷*,杜流云,张中蔚
云南大学软件学院,云南 昆明
收稿日期:2019年9月6日;录用日期:2019年9月20日;发布日期:2019年9月27日

摘 要
染色体是遗传信息载体,正常人的体细胞染色体数目为23对,并有一定的形态和结构。人类从猿到人的长期进化过程中丢失了数万个基因,研究表明,基因中缺失的部分能够反映人类在进化史中的选择,科学家认为DNA的丢失和复制现象可能会是重要的进化动力,研究这些基因变化具有重要意义。人类Y染色体缺失数量较为明显,数百万年前,Y染色体上的基因还有大约1500个之多,而现在却总共只剩下40个。基因缺失属于基因突变的一种,基因突变可以发生在发育的任何时期,在人类中因基因突变引发的疾病有多种,而生物性状的表达由基因控制,从基因序列表达到蛋白质过程中涉及相关的序列有mRNA,ncRNA、cDNA、CDS。本文致力于探索这几种序列之间的关系,以可视化形式将这些基因序列进行可视化。在本文中使用变值概率统计图示表示方法对人类较为特殊的2号染色体、Y染色体、mRNA、CDS、cDNA和ncRNA序列进行分段概率测量,映射成多个2D概率统计图,从而对不同的基因序列特征分布进行比较分析。
关键词 :染色体序列,mRNA,ncRNA,cDNA,CDS,可视化,概率测量

Copyright © 2019 by author(s) and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


1. 引言
目前人类,黑猩猩,大猩猩等灵长类和非灵长类基因组测序逐渐完成,因此人们可以从基因组的角度分析人和其它灵长类、非灵长类之间染色体序列之间的关系。灵长类在进化过程中,一些物种产生了它们特异的染色体重排。几乎所有猿亚目动物都拥有48条染色体,黑猩猩、大猩猩、红毛猩猩无一例外,唯独人类拥有46条染色体。研究发现人类2号染色体是由古猿的两条染色体合并而成的 [1] [2],人类2号染色体上的基因可以和猿类的12 (或称2A)、13 (或称2B)号染色体上的基因一一对应,如图1。
“图1”显示的是使用SYNTENY PORTAL [3] 中的SynCircos将人类的全部染色体基因分布与黑猩猩的全部染色体基因分布进行了一一映射的基因分布图。(a)图是人类染色体上的基因与黑猩猩染色体上基因对应分布关系图。(b)图显示人类2号染色体的基因序列对应分布在黑猩猩的2条染色体2a和2b上。
为何人类在进化过程中出现了染色体合并?基因突变能发生在细胞的各个周期,本文对人类的基因组序列在转录、反转录、翻译过程中的基因序列进行了可视化研究。DNA作为遗传信息载体,维持生物的性状,它是巨大的生物高分子,一般将细胞内遗传信息的携带者枣染色体所包含的DNA总体称为基因组(genome),同一物种的基因组DNA含量总是恒定的,不同物种间基因组大小和复杂程度则差异极大,一般讲,进化程度越高的生物体其基因组构成越大、越复杂。mRNA是由DNA的一条链作为模板转录而来的、携带遗传信息能指导蛋白质合成的一类单链核糖核酸。CDS (Coding DNA Sequence)是编码一段蛋白产物的序列,且该序列中间不含其它非该蛋白质对应的序列,不考虑mRNA加工等过程中的序列变化,与蛋白质的密码子完全对应。miRNA、lncRNA、circRNA等ncRNA (Non-coding RNA)属于表观遗传调控因子,通过与基因、mRNA、蛋白等相互作用,可以在基因表达调控的各个层面调节基因的表达与功能 [4] [5]。近年研究发现,ncRNA在细胞增殖分化、凋亡、肿瘤发生发展过程中都扮演着重要的角色 [6]。cDNA (complementary DNA)是指从mRNA反转录而得到的DNA,是mRNA的一个可靠的拷贝。反转录在生物学上具有重大意义,它对分子生物学的中心法则进行了修正和补充;帮助在致癌病毒的研究中发现致癌基因;它有助于基因工程的实施。DNA,CDS,mRNA,ncRNA,cDNA之间的关系“见图2”。

Figure 1. Circos maps correspond to the chromosome sequences of humans and chimpanzees. (a) (b) from http://bioinfo.konkuk.ac.kr/synteny_portal/htdocs/synteny_circos.php
图1. Circos图对应于人类和黑猩猩的染色体序列。(a) (b)来源于http://bioinfo.konkuk.ac.kr/synteny_portal/htdocs/synteny_circos.php
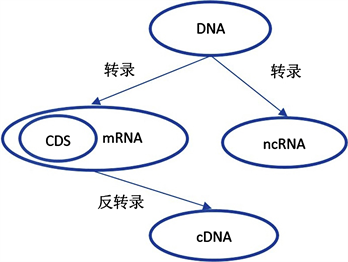
Figure 2. Diagram of the relationship between DNA, mRNA, cDNA and CDs
图2. DNA,mRNA,cDNA,CDs之间的关系图
尽管基因测序技术的迅速发展给基因数据库带来了巨大的数据量,但是计算机的发展也使处理大量基因数据变得容易,对比算法处理全基因组序列时受限于时间复杂度,所以非对比序列分析方法应运而生。用图示或数值表示生物序列,通过生物序列对应的图形或数值间的关系,进而比较生物序列的关系是非对比分析方法的其中一类。将生物序列通过某种映射关系映射为图形可以处理大量的基因序列,生物序列的数值表示主要用数学方法,图示表示方法使人们分析基因序列更直观。与传统图形学不同的是本文研究重点更加侧重于通过可视化图示呈现数据中隐含的信息和规律,本文基于概率论统计方法对人类基因序列进行二维可视化,揭示基因序列中各个碱基之间的一种分布关系。同时也列举了不同测量情况下基因序列可视化结果。
目前生物序列可视化模型方法有很多,并且主要集中工作在DNA序列上,尽管很多可视化模型取得了较好的效果 [7],但是都有其缺陷,例如DNA序列光谱型二维可视化模型,此模型通过将DNA序列转化为二维的曲线,实现了DNA序列的可视化。虽然该模型对于较短的DNA序列可以反映出一些DNA序列的性质,但是该模型并不适合长DNA序列的可视化 [8]。DNA序列双向量二维可视化模型也是一种较常用的DNA序列可视化模型。这种模型采用了DNA行走技术,将4种碱基编码成两个方向的移动向量,但是随着DNA序列长度的增加,整个曲线只是一种趋势,会造成部分细节信息的遗失。因此会引起人眼可能对部分重要信息的忽略。本文引用了变值概率统计可视化方法 [9] [10] [11],此模型方法用于对心电数据处理能够快速、方便、简洁、直观地描述出正常心电和异常心电的分布特性 [12],用于处理编码DNA序列与非编码DNA,得到了很好的低等生物以及高等生物编码区以及非编码区基因特征分布图示 [13],并且此方法应用于生物全基因组序列可视化显示出了生物的特征 [14] [15]。本文首先分析测量了目前DNA序列可视化方法的现状;其次对处理基因序列整体框架与测量序列的模型进行了介绍;最后运用上述基础的测量模型对人类基因序列进行了可视化图示展示分析。
本文的结果如下:
1) 将一整条染色体序列中碱基分布情况展示出来;
2) 将基因序列通过概率统计数值化,对展示出对应关系进行分析;
3) 每条染色体DNA序列之间及cDNA、ncRNA、CDS、mRNA序列之间点聚集分布存在差异性。
2. 常见的DNA序列可视化方法简介
1) 基于分形的DNA序列可视化
郝柏林院士等提出了另一种基于DNA子序列出现频率的可视化方法(简称Hao方法),产生类似的分形图像。这些分形图像充分表明DNA序列具有整体和局部的结构性和长程相关性 [16]。2000年,Daniel Ashlock提出并研究了新的基于迭代函数系统的DNA序列分形表示方法,并引入演化计算思想,对混沌自动机进行演化以对序列进行可视化分类。分形这种可视化所生成图像中的像素点与DNA序列有很好的对应关系,因此分形图像的数学特征就是DNA序列的潜在特征。如果DNA序列分形图像相似,也就表明DNA序列相似。
2) GC含量的一种可视化方法
基因组GC含量的可视化表示方法是通过计算机图像将基因组序列中GC含量的分布与变化情况直观地显示出来。具体是通过滑动窗口在基因组序列中按照一定的规则滑动,将基因组序列分成一系列的小片断。然后对每个片断分别计算得到GC含量。最后将这些片断的GC含量值以条形图表示出来 [17]。此方法对GC含量在基因组中的变化有着非常强大的表现力,但是只是单一表现出GC含量从而忽视了AT碱基的一些特征分布。
3) 基于灰度图像的DNA序列可视化
基于灰度图像的DNA序列可视化模型是由封海清 [18] 等提出的,模型采用了传统的碱基编码方式,将碱基处理成8位二进制数据,再利用成熟的图像处理技术将一维的DNA数据压缩成二维图像,以达到可处理庞大数据的可视化结果。为了更好的突出基于灰度图像的可视化模型在DNA序列可视化上的的优点,作者又引入了熵的概念,熵值与图像无序度呈正比例关系。该方法不存在信息丢失问题,可以处理数据量大的DNA序列,但是基于灰度图像可视化效果区分度并不是那么好。获取有用信息并不那么理想。
4) 彩色的DNA序列可视化模型——Color5
Color5可视化方案是将四个基本字母(A, C, G, T)分别指派给红,黄,蓝,绿四种颜色。用小方块填充不同的颜色来表示DNA碱基一个小方块表示一个碱基,长度为n的碱基序列就取n个小方块。把小方块合并成大方块。大方块边长 ,其中ceil为向上取整数 [8]。其实现方案:
第一步:根据DNA序列的长度,算出大方块所需的边长k。
第二步:建立由k × k个小方块组成的大方块,并把它们都填上白色。
第三步:以Square(1, 1)开始,Jl匾时针标记小方块。
第四步:根据标记,以红,黄,蓝,绿四种颜色填充小方块。
Color的特点没有退化、没有信息丢失、稠密可视化,同时增加了两大优点:彩色的,更便于观察;方的,可以转换为数字矩阵,更方便提炼数字特征。但是在Coloer5没有反映出碱基之间的对应特征分布。
3. 本文模型和方法
处理染色体基因序列的整体框架结构如“见图3”:

Figure 3. Architecture diagram
图3. 体系结构图
3.1. 测量模型
1) ,;S代表一条全序列。
2) m表示S序列分成子序列的分段长度。
3) ,; 是代表四个碱基的字符。
4) 将s序列处理成01序列 ,,;
5) 将Q按照序列长度为m进行分段,分段序列表示为段 ,,;
6) 统计数量 ,,;
7) 非归一化概率测度 ;
8) 归一化概率测度 ;
9) 统计非归一化概率数量 ,,;
10) 统计归一化概率数量 ,, 。
3.2. 映射模型
本文中用热力图原理实现图示,非归一化图示时以概率测度 作为横纵坐标,实现非归一化图示时 作为横纵坐标。
3.3. 测量原理
从原始的染色体基因序列数据到最终的2D特征分布图主要经过以下步骤:数据分类、数据处理、数据可视化。
灵长类物种中全基因组序列数据中包含常染色体与性染色体,在进行数据处理中直接处理一条染色体序列,每一条染色单体可看作一条双螺旋的脱氧核糖核酸分子,脱氧核苷酸又是由脱氧核糖、磷酸和含氮碱基组成,碱基有4种,分别是腺嘌呤(A)、鸟嘌呤(G)、胞嘧啶(C)和胸腺嘧啶(T),故可将一条染色体基因序列看做是由A、G、C、T四个字符连接而成的字符串,可表示为: ,,序列长度用N表示,将序列按长度为m分成k个子序列段。分别统计出每个子序列中A (腺嘌呤),G (鸟嘌呤),C (胞嘧啶),T (胸腺嘧啶)的个数,分别用CA,CG,CC,CT,来表示其对应的个数。
以N表示的测量参数为基础,进行概率测度对应的统计,概率测度分为2种,归一化百分比和非归一化百分比 [11]。非归一化测度:碱基个数除以子序列长度得到百分比;归一化测度:碱基个数除以两种互补碱基的个数。每个碱基的归一化测度对应计数均在0至1之间的百分比,将该数据作为坐标位置映射部分的输入,按照一定的规则,对其进行处理,最终得到每个点的横纵坐标 [13]。
4. 可视化结果
本文用采用热力图实现原理对概率统计所得到数值进行可视化图示,以特殊高亮的形式表示出点的聚集程度密集。图中Z表示点的密集程度对应的色彩变化,数量由低到高——色彩由暗到特殊高亮。图中深红部分点的聚集度最高,深蓝部分点聚集最少。本文所得图示中心部分是点分布最密集的区域,然后逐渐向边缘扩散点聚集密度逐渐降低。
用较灵长类的染色体序列,通过控制可控参数下形成二维特征分布图,通过控制横纵坐标的变量及其分段长度m适度的情况下得到的特征分布图如下:根据四种碱基的不同性质,在三个层面上进行划分:
嘌呤R=A、G/嘧啶Y=C、T;
氨基M=A、C/羰基K=G、T;
强氢键S=G、C/弱氢键W=A、T。
以嘌呤A的概率、G的概率得到“图4”(a1) (b1);
以嘧啶C的概率、T的概率得到“图4”(a2) (b2);
以氨基A的概率、C的概率得到“图4”(a3) (b3);
以羰基G的概率、T的概率得到“图4”(a4) (b4);
以强氢键G的概率、C的概率得到“图4”(a5) (b5);
以弱氢键A的概率、T的概率得到“图4”(a6) (b6)。
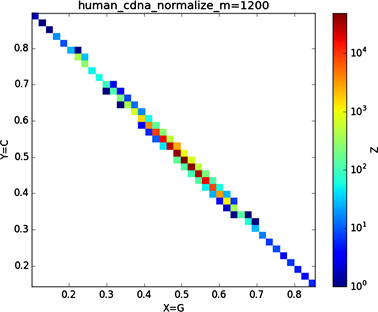
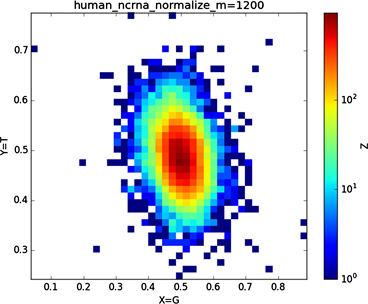
“图4”(选择样品为人类的2号染色体序列)。
非归一化特征分布图:(a1, a2, a3, a4, a5, a6);
归一化特征分布图:(b1, b2, b3, b4, b5, b6)。
由上“图4”所示,分段长度m:1200;
横坐标X:A (腺嘌呤),C (胞嘧啶),A (6-氨基嘌呤),G (4-氨基-2-羰基嘧啶),G,A;
 (a1)
(a1)
 (b1)
(b1)
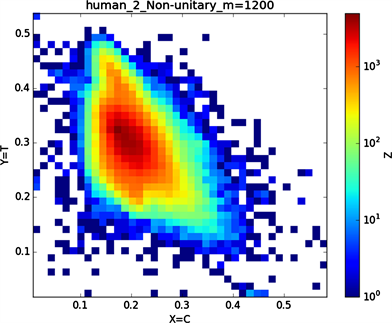 (a2)
(a2)
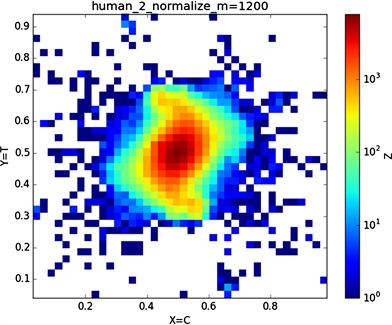 (b2)
(b2)
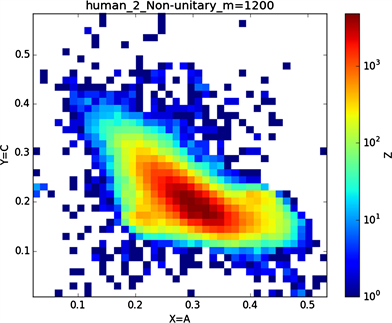 (a3)
(a3)
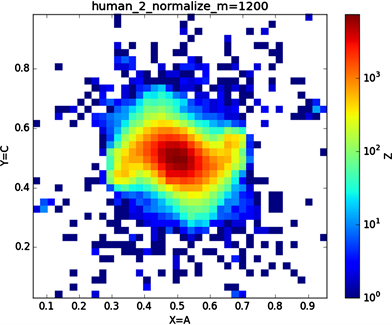 (b3)
(b3)
 (a4)
(a4)
 (b4)
(b4)
 (a5)
(a5)
 (b5)
(b5)
 (a6)
(a6)
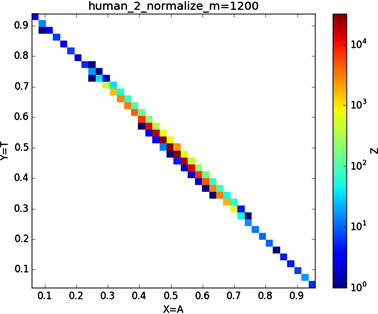 (b6)
(b6)
Figure 4. Normalized and non-normalized graphs of human chromosome 2
图4. 人类2号染色体归一化与非归一化图示
纵坐标Y:G (鸟嘌呤),T (胸腺嘧啶),C (2-氨基-6-羟基嘌呤),T (5-甲基尿嘧啶),C,T;
横纵坐标一一对应形成二维特征分布图。
由“图3”中非归一化与归一化的对比图中会发现非归一化中(a1)与(a2)的特征分布点相对于与(a3)或(a4)或(a5)或(a6)的特征分布点更为相似,(a3)与(a4)的特征分布点相对于(a1)或(a2)或(a5)或(a6)的特征分布点二者特征点更为相似,由此可以看出碱基之间一种分布关系。在进行归一化之后,(b1),(b2),(b3),(b4)的特征分布点都有一定的共性,通过旋转maps会发现这四个特征分布图较为相似;“图3”中非归一化到归一化的特征分布图中我们发现(a5) (b5),(a6) (b6),特征分布变化较大,(a5)特征分布图的横纵坐标对应的是G、C碱基概率,(a6)特征分布图的横纵坐标对应的是A、T碱基概率,可以看到(a5) (a6)的特征分布图是呈对称分布的,我们知道GC,AT它们之间对应的关系是碱基互补配对,从图(a5) (a6)的结果图中我们可以得到的信息是在整条染色体序中如果存在某一区域一个碱基与其互补碱基对应分布概率(g, f),那在这条染色体另外的某一区域一定存在该碱基与其互补碱基的概率分布为(f, g),同理可以解释在进行归一化处理之后得到对应的(b5) (b6)特征分布图图示结果,从(b5) (b6)的特征分布图中显示出碱基与其互补碱基概率分布的一种相关性,在所得到的图示结果中我们可以得到的信息是在染色体序列中一种碱基在某一区域分布概率大时则该碱基的互补碱基在该区域分布数量相对较小。
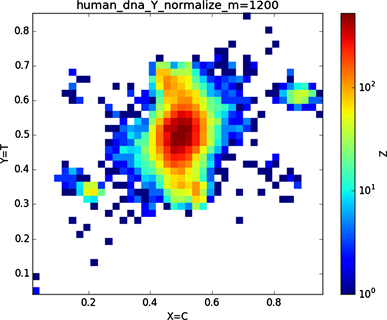
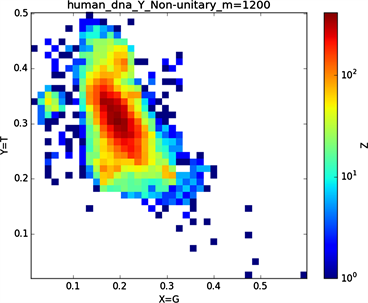
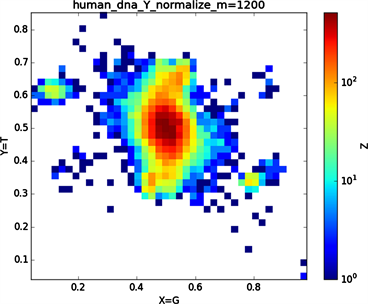
将“图4”与”图5”的图示结果作为对比分析发现其特征分布差异较大,虽同为染色体,Y染色体序列图示的边缘出现较为特殊的聚集点。基因在表达为外在性状时是经过转录翻译等过程,本文将蛋白质表达过程中基因序列进行了可视化如下。
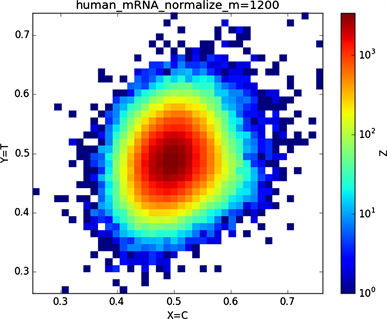
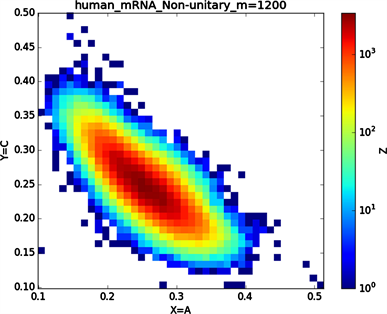
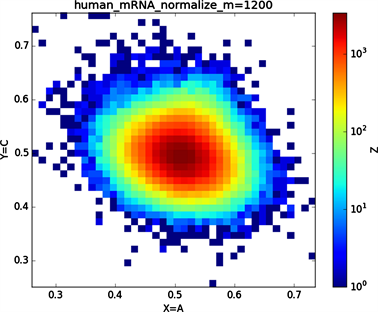
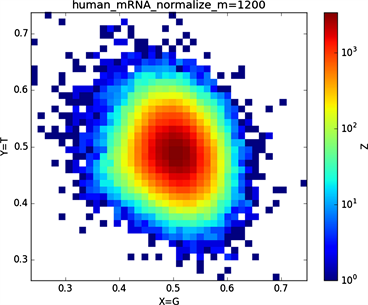
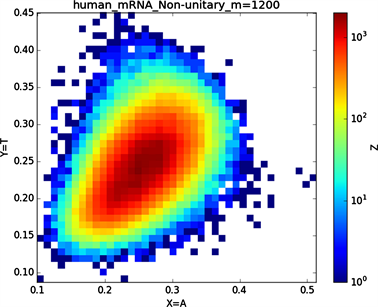
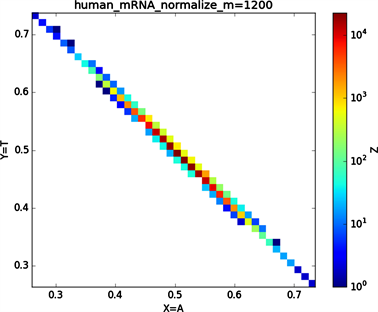
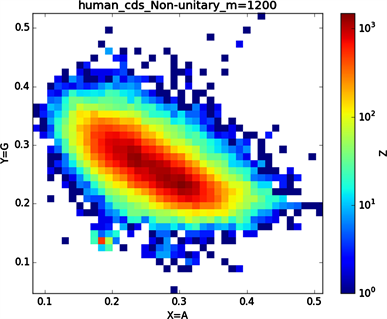
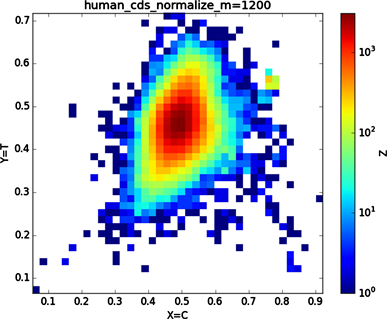
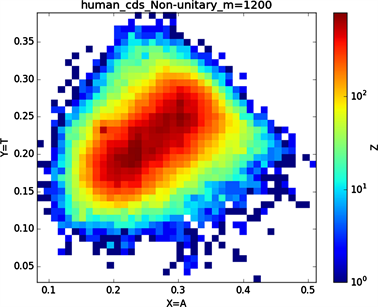
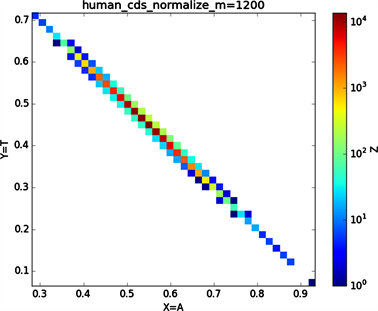
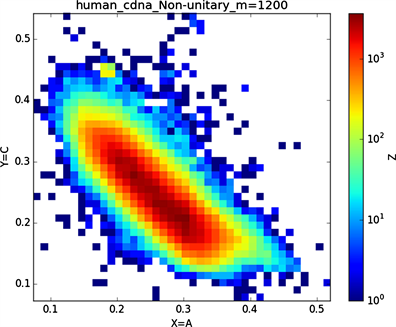
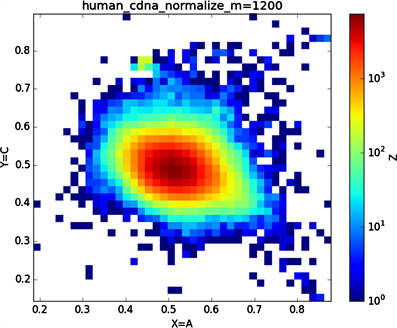
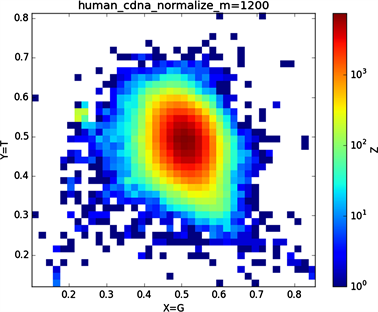
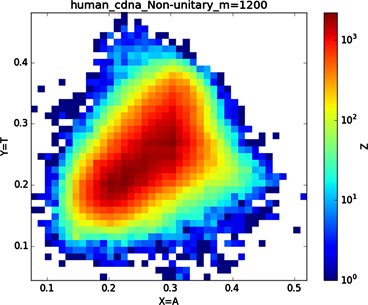
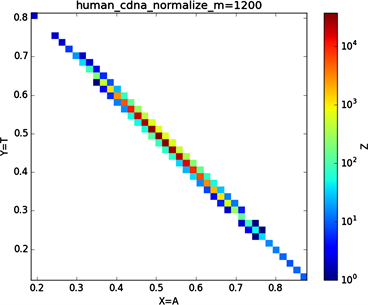
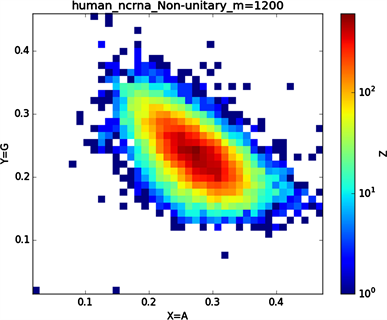
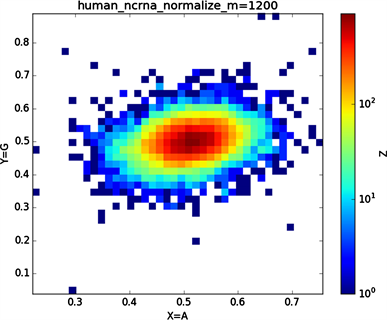
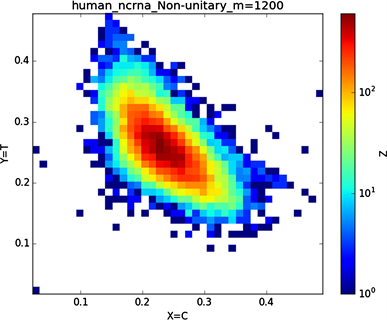
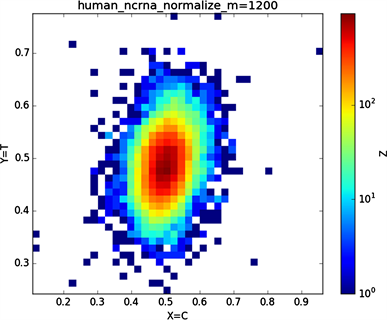
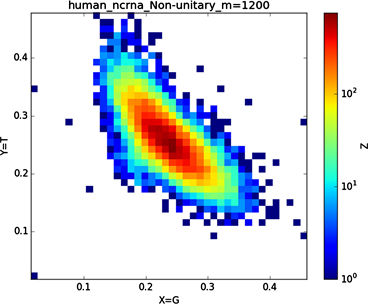
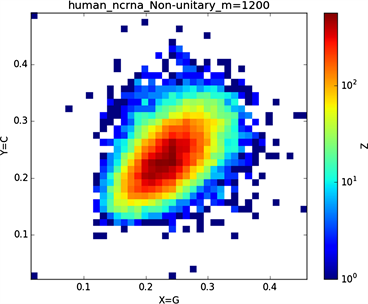
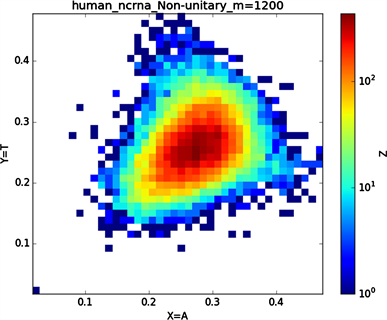
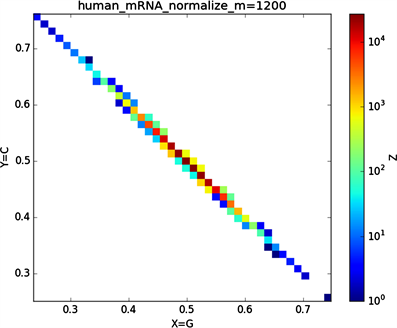
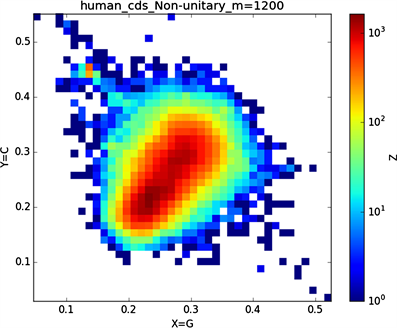
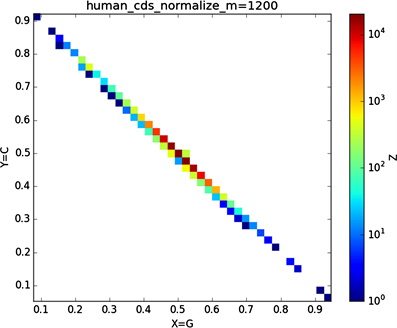
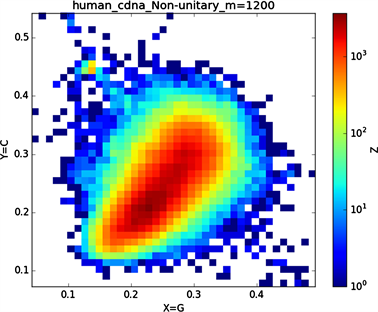
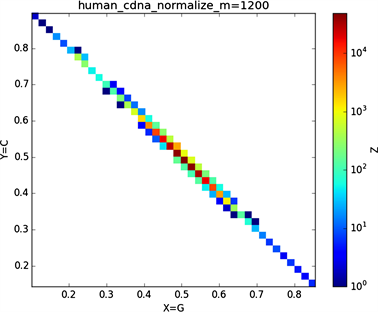
全球性的转录分析发现66%的基因组被转录,其中80%是呈现活性转录的生物化学标记,然而不到2%的能够编码蛋白质 [19]。绝大多数非编码的调控元件被转录成非编码RNAS (non-coding RNAS, RNAS) [20]。从“图6”,“图7”、“图8”与“图9”的图示结果中我们发现“图7”中的CDS图示与“图8”中cDNA分布较为相似,在观察图7与“图8”时我们会发现在图示边缘部分两组图示都出现又聚集的点。从而推进我们对CDS与cDNA之间的联系进行下一步研究。“图9”中非编码RNA和“图6”中mRNA图示特征分布区别较大。mRNA是由DNA的一条链作为模板转录而来的、携带遗传信息能指导蛋白质合成的一类单链核糖核酸。cDNA是反转录的产物,反转录应用于基因工程中的克隆 [21] [22] [23]。真核生物的mRNA或其他RNA的cDNA,在遗传工程方面广为应用。
在“图10”的对比图中,(a1)、(a2)、(a3)、(a4)分别是人类mRNA、CDS、cDNA、ncRNA序列的非归一化对应的图示,(a1)、(a2)、(a3)、(a4)是人类mRNA、CDS、cDNA、ncRNA序列的归一化图示。从图示中我们能观察到两种方法都能体现出每种序列分布存在差异性,(a1)、(a2)、(a3)、(a4)对应的图示能更详细体现序列的分布,(a1)、(a2)、(a3)、(a4)图示则体现出碱基概率分布之间的相关性。两种方法共同结合能更好的对图示结果进行分析。
5. 总结
本文基于概率统计的变值图示方法对人类的染色体序列和人类主要一些序列进行可视化,并将这些序列的图示结果做了比较分析,解决之前一些可视化模型因为序列长而无法观察出全部的碱基分布的一些特征的问题。本文中可视化模型体现的优点是观察者可观察到目前已经测序完成的一整条染色体序列碱基之间的概率分布,图示结果也展示出碱基之间对应的分布关系。在比较人类染色体序列图示与其cDNA、ncRNA、CDS、mRNA序列图示时能够较易观察出它们之间概率特征分布存在一定的异或性,CDS概率特征分布图与cDNA概率特征分布图示相对于mRNA、ncRNA存在更为相似的分布。本文可视化方法不足之处是当基因序列图示分布出现差异时不能确定呈现差异基因序列的具体分布位置;在图示结果中虽然体现出一条染色体序列中一种碱基与其互补碱基分布有着相关性,但是却不能确定碱基与
 (a1)
(a1)
 (b1)
(b1)
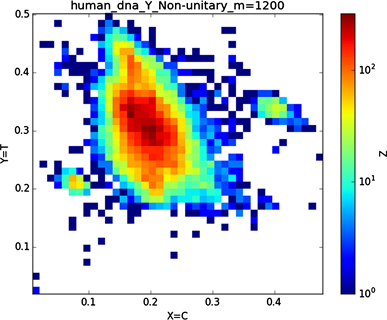 (a2)
(a2)
 (b2)
(b2)
 (a3)
(a3)
 (b3)
(b3)
 (a4)
(a4)
 (b4)
(b4)
 (a5)
(a5)
 (b5)
(b5)
 (a6)
(a6)
 (b6)
(b6)
Figure 5. Non-normalization and normalization of human Y chromosome sequence
图5. 人类Y染色体序列非归一化与归一化图示
 (a1)
(a1)
 (b1)
(b1)
 (a2)
(a2)
 (b2)
(b2)
 (a3)
(a3)
 (b3)
(b3)
 (a4)
(a4)
 (b4)
(b4)
 (a5)
(a5)
 (b5)
(b5)
 (a6)
(a6)
 (b6)
(b6)
Figure 6. Chart of human mRNA sequence
图6. 人类mRNA序列图示
 (a1)
(a1)
 (b1)
(b1)
 (a2)
(a2)
 (b2)
(b2)
 (a3)
(a3)
 (b3)
(b3)
 (a4)
(a4)
 (b4)
(b4)
 (a5)
(a5)
 (b5)
(b5)
 (a6)
(a6)
 (b6)
(b6)
Figure 7. Human CDS sequence diagram
图7. 人类CDS序列图示
 (a1)
(a1)
 (b1)
(b1)
 (a2)
(a2)
 (b2)
(b2)
 (a3)
(a3)
 (b3)
(b3)
 (a4)
(a4)
 (b4)
(b4)
 (a5)
(a5)
 (b5)
(b5)
 (a6)
(a6)
 (b6)
(b6)
Figure 8. Human complementary DNA sequence map
图8. 人类complementary DNA序列图示
 (a1)
(a1)
 (b1)
(b1)
 (a2)
(a2)
 (b2)
(b2)
 (a3)
(a3)
 (b3)
(b3)
 (a4)
(a4)
 (b4)
(b4)
 (a5)
(a5)
 (b5)
(b5)
 (a6)
(a6)
 (b6)
(b6)
Figure 9. Chart of Homo sapiens non-coding RNA sequence
图9. 人类非编码RNA序列图示
 (a1)
(a1)
 (b1)
(b1)
 (a2)
(a2)
 (b2)
(b2)
 (a3)
(a3)
 (b3)
(b3)
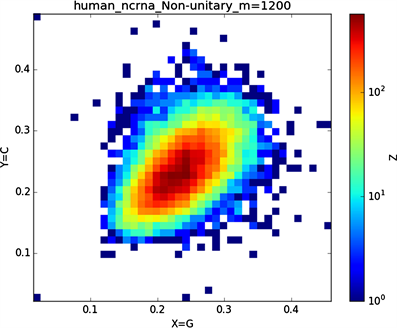 (a4)
(a4)
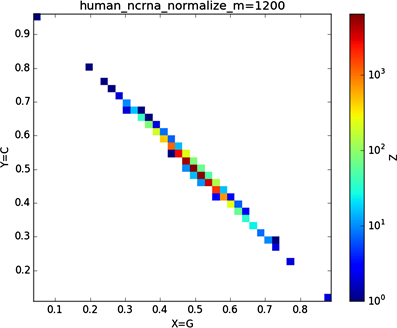 (b4)
(b4)
Figure 10. Comparison of non-normalized and normalized sequences of human mRNA, CDS, cDNA and ncRNA
图10. 人类的mRNA、CDS、cDNA、ncRNA序列非归一化与归一化对比图
其互补碱基之间在同一区域存在相关性关系。希望文中给出的可视化方法,提出的分析测量模型以及图示中所展示的染色体序列碱基之间对应的概率分布特征(提取的测量特征可视化机制)、cDNA及其CDS等序列图示结果分布特征能为后续基因工程中基因数据以及结构的可视化分析的应用研究提供坚实的模型和实践基础。
致谢
感谢云南大学软件学院,感谢云南省软件工程重点实验室提供良好的工作环境。感谢国家自然科学基金(K1020720)和中国云南省海外高级学者项目(W8110305)和中国云南省科技计划项目(KC1810123)提供了对该项目的资金支持。
文章引用
郑华仙,郑智捷,杜流云,张中蔚. 人类基因序列二维可视化分析
Two-Dimensional Visualization Analysis of Homo Sapiens Gene Sequences[J]. 计算生物学, 2019, 09(03): 59-80. https://doi.org/10.12677/HJCB.2019.93009
参考文献
- 1. 刘笑麟. 人类进化, 基因大流失[J]. 大科技: 科学之谜(A), 2018(4): 46-47.
- 2. 晏子悠. 谁偷了我的染色体[J]. 大科技(科学之谜), 2007(2): 42-43.
- 3. http://europepmc.org/articles/PMC4987893
- 4. 潘书贤, 周光明, 胡文涛. 非编码RNA参与昼夜节律调控研究进展[J]. 航天医学与医学工程, 2019, 32(2): 178-182.
- 5. 杜娟, 陈亚妮, 史海燕, 吴博, 王爱红, 殷松娜. 长链非编码RNA LINC00519在胃癌中的表达及其临床意义[J/OL]. 山西医科大学学报, 2019(4): 484-488.
- 6. Li, W., Wang, R., Ma, J.Y., et al. (2017) A Human Long Non-Coding RNA ALT1 Controls the Cell Cycle of Vascular Endothelial Cells via ACE2 and Cyclin D1 Pathway. International Journal of Experimental Cellular Physiology Biochemistry & Pharmacology, 43, 1152-1167. https://doi.org/10.1159/000481756
- 7. 解小莉. 生物序列的分析方法及其进化模型研究[D]: [博士学位论文]. 杨凌: 西北农林科技大学, 2012.
- 8. 张柱金. DNA序列二维可视化研究[D]: [博士学位论文]. 武汉: 华中科技大学, 2011.
- 9. Zheng, J. (2018). Variant Construction from Theoretical Foundation to Applications. Springer, Ber-lin.
- 10. 郑智捷. 在变值测量模拟中的条件概率统计分布[J]. 光子学报, 2011, 40(11): 1662-1666.
- 11. 完竹, 郑智捷. DNA序列一维分段测量分布可视化[J]. 云南大学学报(自然科学版), 2013, 35(S2): 1-6.
- 12. 吉艳. 基于变值测量的心电数据序列可视化应用研究[D]: [硕士学位论文]. 昆明: 云南大学, 2016.
- 13. 刘玉倩, 郑智捷. 编码和非编码DNA序列的可视化分析[J]. 计算生物学, 2014, 4(2): 20-31.
- 14. Mao, Y., Zheng, J. and Liu, W. (2017) Mapping Whole DNA Sequence on Variant Maps. Proceedings of the 2017 IEEE/ACM International Conference on Advances in Social Networks Analysis and Mining, Sydney, 31 July-3 August 2017, 1037-1040. https://doi.org/10.1145/3110025.3110140
- 15. 刘文嘉, 郑智捷. DNA序列互补匹配特征分析及可视化系统[J]. 计算生物学, 2015, 5(4): 49-57.
- 16. 王树林, 王戟, 陈火旺, 张波云. 基于分形的DNA序列可视化表示研究[J]. 计算机科学, 2006(7): 158-163.
- 17. 王安慧. 基因组信息的计算机可视化若干关键技术研究[D]: [博士学位论文]. 沈阳: 东北大学, 2010.
- 18. 封海清, 陆祖宏. 一种新型的基于图像的DNA序列可视化模型[J]. 生物信息学, 2014, 12(2): 133-139.
- 19. Djebali, S., et al. (2012) Landscape of Transcription in Human Cells. Nature, 489, 101-108.
- 20. 李平, 庞智, 朱剑云, 孙琛琛, 孙康云. 非编码RNA对冠心病发生发展影响的研究进展[J/OL]. 医学综述, 2019(8): 1474-1479. http://kns.cnki.net/kcms/detail/11.3553.r.20190419.1033.008.html, 2019-05-05.
- 21. 崔薇, 陈楠, 苗震, 李和平, 刘伟石, 夏彦玲. 梅花鹿PTN基因cDNA克隆及表达分析[J]. 黑龙江畜牧兽医, 2019(7): 129-132.
- 22. 孙盛明, 傅洪拓, 宣富君, 戈贤平, 朱健, 吴旭干. 青虾C型凝集素结构域家族3的cDNA克隆、原核表达和定位[J/OL]. 水产学报, 1-14. http://kns.cnki.net/kcms/detail/31.1283.s.20190408.0929.012.html, 2019-04-30.
- 23. 何鹏, 江世贵, 李运东, 杨其彬, 姜松, 杨丽诗, 黄建华, 周发林. 斑节对虾GLUT1基因cDNA的克隆与表达分析[J]. 南方水产科学, 2019, 15(2): 72-82.
NOTES
*通讯作者。