Modern Physics
Vol.4 No.05(2014), Article ID:14131,8 pages
DOI:10.12677/MP.2014.45014
Electronic Structures and Optical Properties of the Oxygen-Deficient SrTiO3 from First-Principles Calculation
Department of Physics, Yunnan University, Kunming
Email: *yhe@ynu.edu.cn
Copyright © 2014 by authors and Hans Publishers Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received: Sep. 1st, 2014; revised: Sep. 8th, 2014; accepted: Sep. 15th, 2014
ABSTRACT
In recent years, perovskite oxides attracted widely attention due to its unique structure and the chemical and physical properties. SrTiO3 (hereinafter referred to STO) is a kind of typical perovskite oxides. It has the characteristics of typical perovskite structure, and its high dielectric constant, low dielectric loss and good thermal stability made it easier to attract more attentions. In this paper, we investigate the electronic and optical properties of STO using LDA + U method. We found that this method predicts more accurate band gap for STO. The oxygen vacancy induced local defect state and new absorption band, which enhanced the efficiency of absorption in the visible region.
Keywords:First-Principles Calculation, SrTiO3, Defect, Electronic Structure, Optical Properties

具有氧缺陷的SrTiO3的电子结构和光学性质的第一性原理研究
陈清源,何 垚*
云南大学物理科学技术学院,昆明
Email: *yhe@ynu.edu.cn
收稿日期:2014年9月1日;修回日期:2014年9月6日;录用日期:2014年9月15日

摘 要
近年来,钙钛矿型氧化物由于其具有独特的结构以及丰富的物理化学性质而受到广泛的关注。SrTiO3(以下简称STO)是一种典型钙钛矿型氧化物,具有典型钙钛矿结构所具有的特点,并且其较高的介电常数、低介电损耗及良好的热稳定性使其备受关注。本文针对其结构特性及光学性质运用了第一性原理进行了研究,发现运用LDA + U方法能够更准确地描述其电子结构,获得与实验值更加吻合的禁带宽度。在这基础上,我们能够准确地预言,通过氧缺陷能够引入缺陷态,并在吸收谱中引入新的吸收带,实现对可见光的有效利用。
关键词
第一性原理计算,SrTiO3,缺陷,电子结构,光学性质

1. 引言
SrTiO3(下简称STO)具有丰富而特殊的特性,STO具有典型的钙钛矿型结构所具备的特点,使其成为了一种具有广泛用途的电子功能陶瓷材料,它的优点有介电常数高、介电损耗低、热稳定性好等。在电子、机械和陶瓷工业上有着广泛地应用,在室温条件下,SrTiO3属于立方晶系,是一种典型的ABO3型钙钛矿复合氧化物。
STO晶体是一种具有宽禁带的半导体材料(带隙为3.25 eV [1] [2] ),光催化活性很好,并具有独特的电磁性质和氧化还原催化活性,被广泛的应用在光催化分解水制氢、光催化降解有机污染物和光化学电池等光催化领域。但是其较大带隙(3.25 eV)导致其只能吸收紫外光,限制了其对太阳光的有效利用。利用其产生的缺陷减小其带隙是使其具有可见光响应的有效手段,在STO中的氧缺陷可以形成亚带隙能级,亚带隙能级可以更多的响应可见光,更多的利用可见光能量,提高其光子利用率。
本文运用第一性原理计算对STO进行模拟计算,在计算过程中我们采用LDA + U [3] 方法对带隙进行修正。在此基础上对其电子结构以及光学性质进行研究。并本着提高其光子利用率为目标,重点研究氧缺陷时SrTiO2.875的光学性质。
2. 模型
本文所有计算由VASP软件包完成[3] [4] 。VASP是基于密度泛函理论的第一性原理量子力学程序。在密度泛函理论框架下,采用平面投影缀加波(projector augmented wave,PAW)处理价电子的相互作用[5] [6] ,电子关联相互作用采用基于广义梯度近似(GGA)的PBE泛函[1] 。本文将Sr的4p65s2,Ti的3p63d24s2,O的2s22p4电子视为价电子来处理。所有计算中选取平面波截断能为400 eV。对原子结构优化的标准为原子间的作用力不大于0.01 eV/Å。
无缺陷的STO晶体属于Pm3m空间群,为立方晶系,实验晶格常数为a = 0.39051 nm。如图1所示,在一个原胞中包含一个Ti原子,一个Sr原子和三个氧原子。其中Ti位于立方原胞中心,Sr位于立方顶点,O位于面心。本文经过原子结构弛豫后得到的晶格常数为a = 0.3905 nm,与试验吻合得比较好。

Figure 1. The cell structure of perfect STO
图1. 无缺陷STO单胞结构
3. 结果与讨论
3.1. 具有氧缺陷的STO电子结构及光学性质的研究
在LDA + U方法下无缺陷单胞STO的禁带宽度和电子态密度都和实验值吻合很好,由此证明我们采用的LDA + U方法得当。且在STO的缺陷中,氧缺陷是最容易出现和存在的,所以下面我们只着重研究氧缺陷态STO的电子结构和光学特性。
为了减小缺陷带来的分子内部的相互作用,使计算更为准确,我们采取了如图2的2 × 2 × 2超晶胞结构进行研究。
采用2 × 2 × 2的包含40个原子的超晶胞模拟无限大的晶体,通过移去一个氧的办法产生相应的空位,形成SrTiO2.875体系。优化过程中选用paw-pbe赝势方法,截断能取400 eV,进行离子弛豫时,收敛标准为原子间作用力不大于0.01 eV/Å,布里渊区积分采用Monkhorst-pack型的k点网格。采用的高对称K点走向为X→R→M→G→R。
3.1.1. LDA + U方法下SrTiO3电子结构研究
首先用LDA + U方法计算无缺陷SrTiO3 (2 × 2 × 2)总态密度和能带图,如图3所示。
我们选取实验时的晶格常数a = 0.3905 nm,并用LDA + U方法进行计算,近似方式选择Dudarev近似方式。其中J = 0.64,并选取不同的U值对LDA + U方法进行测试[6] [7] 。随着U值得改变,带隙宽度接近实验值,带隙宽度为3.27 eV。与实验3.25 eV符合的比较好。故下面我们以此为基础来进行进一步分析。
下面我们将和具有氧缺陷时的能带结构作比较。
3.1.2. LDA + U方法下SrTiO2.875电子结构研究
我们从STO的电子结构出发,首先计算了有氧缺陷的SrTiO2.875 (2 × 2 × 2)态密度及能带图以及分波态密度,见图4-6。
由以上几幅态密度和能带图我们可以分析得到:
1) 与无缺陷的STO相比,氧缺陷的存在很明显的在禁带中出现了缺陷能级,它清楚地表现在能带图和态密度图中的0 eV附近。
2) SrTiO2.875体系的导带底主要是由Ti3d电子占据,价带顶主要由02p电子占据。
3) 由于O空位的存在,Ti的分波态密度在导带底费米能级附近有较大的变化。SrTiO2.875体系中费米能级进入了导带,O空位的存在使STO由绝缘性变成n型半导体。


Figure 2. The supercell structure of 2 × 2 × 2 STO: left is perfect STO model and right is SrTiO2.875 model
图2. 左图是perfect无缺陷模型 右图是氧空位模型
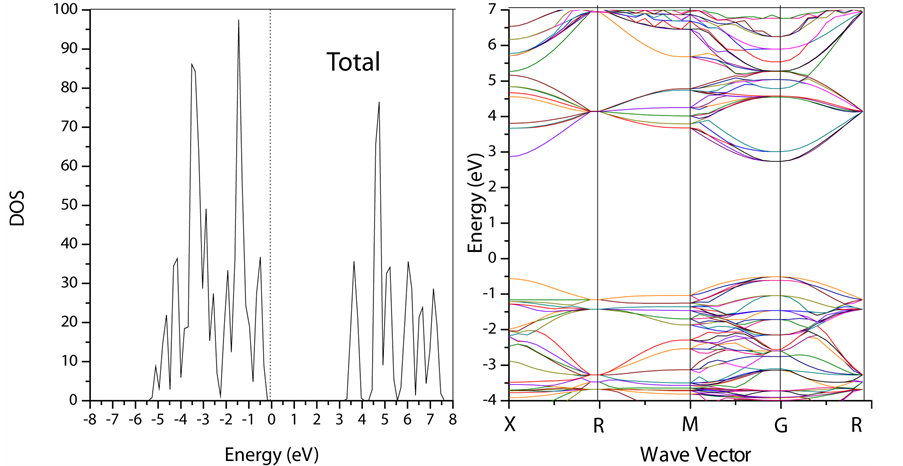
Figure 3. Density of states and band structure figure of perfect 2 × 2 × 2 STO supercell’s figure by LDA + U method
图3. LDA + U方法下SrTiO3 (2 × 2 × 2)的总态密度以及能带
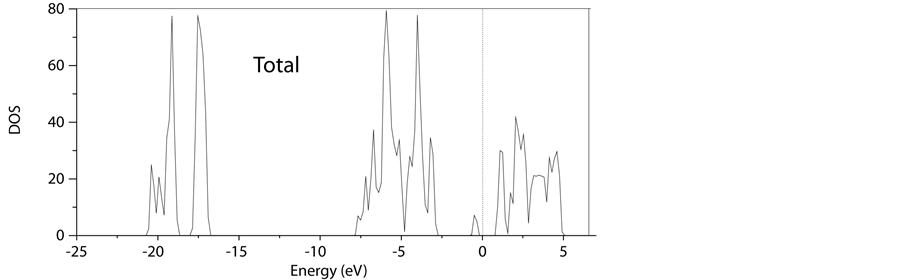
Figure 4. Density of states of defect 2 × 2 × 2 SrTiO2.875 supercell’s figure by LDA + U method
图4. LDA + U方法下SrTiO2.875 (2 × 2 × 2)的总态密度
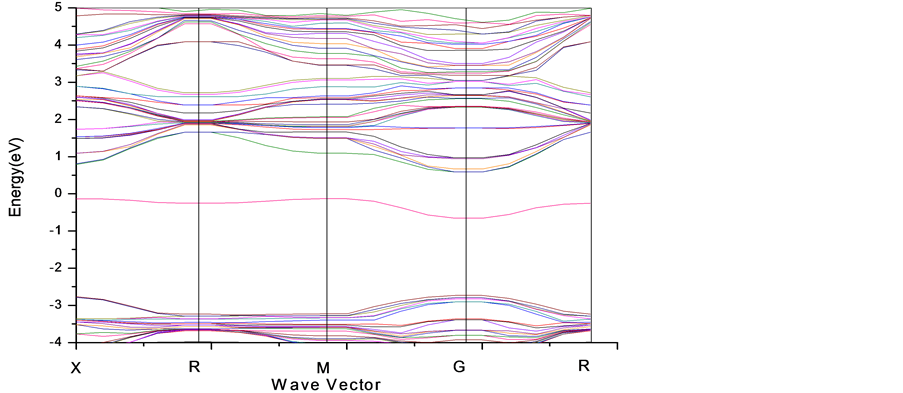
Figure 5. Band structure of defect 2 × 2 × 2 SrTiO2.875 supercell’s figure by LDA + U method
图5. LDA + U方法下SrTiO2.875 (2 × 2 × 2)的能带

Figure 6. Partial density of states of O defect STO’s figure by LDA + U method
图6. LDA + U方法下SrTiO2.875 (2 × 2 × 2)的分波态密度
4) 费米能级下方的缺陷能级主要由Ti原子的3d态电子占据,同时Ti的4s,3p态也有一定影响;氧原子2p态电子也有少量占据缺陷能级,但是贡献都较小,可以忽略[8] -[12] 。
3.2. LDA + U方法对STO光学性质的研究
3.2.1. LDA + U方法对无缺陷STO光学性质的研究
首先我们用LDA + U方法研究2 × 2 × 2的无缺陷的STO的光学性质,见图7。
图7左边是LDA + U方法研究的STO介电函数图,可以看出:当频率为零或很低时,介电函数的虚部 ,静态介电常数
,静态介电常数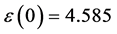 ,随着频率增加,介电函数取复数形式(虚部不为0),对于STO来说,能量大于能隙3.27
eV后
,随着频率增加,介电函数取复数形式(虚部不为0),对于STO来说,能量大于能隙3.27
eV后 。
。
右图则是吸收系数与光子能量之间的关系曲线。从图可以看出,当光子能量在0~3.27 eV之间时,STO几乎不存在吸收;即STO仅仅只对紫外光有吸收,而对可见光没有吸收利用,其对光子的利用率较低,正是基于这样的结果我们下面引入氧缺陷,目的在于提高STO对于光子的利用率。
3.2.2. LDA + U方法对氧缺陷STO光学性质的研究
下面我们用LDA + U方法研究2 × 2 × 2的具有氧缺陷STO的光学性质,见图8。
可以分析得到:
图8(a):LDA + U方法研究的SrTiO2.875介电函数图,可以看出当频率为零或很低时,介电函数的虚部 ,静态介电常数
,静态介电常数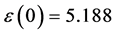 ,随着频率增加,
,随着频率增加,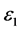 也快速增加,同时在1.47 eV处达到第一个峰值,介电函数取复数形式(虚部不为0),虚部
也快速增加,同时在1.47 eV处达到第一个峰值,介电函数取复数形式(虚部不为0),虚部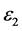 代表介质损耗,它是由于分子的电极化过程跟不上外场变化而引起的。对于SrTiO2.875来说,能量大于0.98
eV后
代表介质损耗,它是由于分子的电极化过程跟不上外场变化而引起的。对于SrTiO2.875来说,能量大于0.98
eV后 ,SrTiO2.875的吸收边为0.98 eV。介电函数虚部的主要的峰值出现在1.47
eV,3.43 eV,4.91 eV,7.86 eV,9.33 eV和22.11 eV,结合能带图和分波态密度图可以看出,第一个峰和第二个峰是缺陷能级的Ti3d和O2p电子向导带的跃迁,第三主要价带顶电子O2p电子到缺陷能级的跃迁,第四到第五个主要是O2p电子到导带Ti3d的跃迁,最后一个峰主要是Sr4p和O2s电子到缺陷能级的跃迁所致。由图可以看出它在低能量时多出了两个峰值。
,SrTiO2.875的吸收边为0.98 eV。介电函数虚部的主要的峰值出现在1.47
eV,3.43 eV,4.91 eV,7.86 eV,9.33 eV和22.11 eV,结合能带图和分波态密度图可以看出,第一个峰和第二个峰是缺陷能级的Ti3d和O2p电子向导带的跃迁,第三主要价带顶电子O2p电子到缺陷能级的跃迁,第四到第五个主要是O2p电子到导带Ti3d的跃迁,最后一个峰主要是Sr4p和O2s电子到缺陷能级的跃迁所致。由图可以看出它在低能量时多出了两个峰值。
折射率N = n + ik 与ε之间存在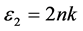 ,
, 的关系。从图8(a)和图8(b)对应曲线来看,静态折射率为2.27,且在1.47 eV,3.43
eV处的峰值为缺陷所引起的。
的关系。从图8(a)和图8(b)对应曲线来看,静态折射率为2.27,且在1.47 eV,3.43
eV处的峰值为缺陷所引起的。
反射率与折射率n、k之间的关系如下: ,从图8(c)可看出,光子能量在0~0.98
eV之间,k接近0,R主要是由静态折射率引起的。R的第一个峰值出现在1.47 eV,R在1.47 eV和3.43 eV出现的峰值对应的n的峰值出现在1.47 eV和3.43
eV,此时反射率增加是缺陷带吸收了部分波长的光引起的[13] -[18] 。
,从图8(c)可看出,光子能量在0~0.98
eV之间,k接近0,R主要是由静态折射率引起的。R的第一个峰值出现在1.47 eV,R在1.47 eV和3.43 eV出现的峰值对应的n的峰值出现在1.47 eV和3.43
eV,此时反射率增加是缺陷带吸收了部分波长的光引起的[13] -[18] 。
为了更好地观察吸收系数,我们将上图d进行放大,见图9。
在吸收谱图9中可以看出该图是吸收系数与光子能量之间的关系曲线。当光子能量在0~0.98 eV之间时,具有氧缺陷的STO几乎不存在吸收;光子能量在0.98~3.93 eV,吸收系数的极大值出现在1.47 eV、3.43 eV附近。1.47 eV,3.43 eV处的峰值为缺陷所引起的。结合图8(a),可以知道这两个吸收峰是由缺陷能级Ti3d和O2p的电子向导带的跃迁引起的,而第三个4.91 eV处的峰是由价带顶O2p电子到缺陷能级的跃迁引起的,在3.93~15.23 eV之间的能量范围时,材料又出现明显的吸收过程,这个过程与无缺陷的分析基本一致,由此可知缺陷能级可以使得STO除了吸收紫外光还能更多的响应可见光,更多的利用可见光,使光子利用率提高。
4. 结论
本工作根据第一性原理运用LDA + U方法研究了STO材料的电子结构以及光学性质,对其能带、态密度、介电函数、折射率、反射率、吸收系数进行了分析。计算并得到了与实验值吻合的结果。并着重研究了具有氧缺陷的STO材料的电子结构和光学特性,发现无缺陷的STO只能响应紫外光,而具有氧缺陷的SrTiO2.875能够响应可见光。得到了氧缺陷的STO可以更多的响应可见光,更多的利用可见光能量,使光子利用率提高的结论。例如在基于STO的光催化分解水制氢、光催化降解有机污染物和光化学电池等光催化领域,可以通过在STO中引入氧缺陷提高光催化分解制氢效率、光催化降解效率及光电转化效率等。

Figure 7. The left figure black and red lines represent our calculated real and imaginary parts of dielectric function ε(ω) respectively of perfect STO (2 × 2 × 2) by LDA + U method. The right figure is the adsorption coefficient I of perfect STO (2 × 2 × 2) by LDA + U method
图7. LDA + U方法下纯STO (2 × 2 × 2)的复介电函数图
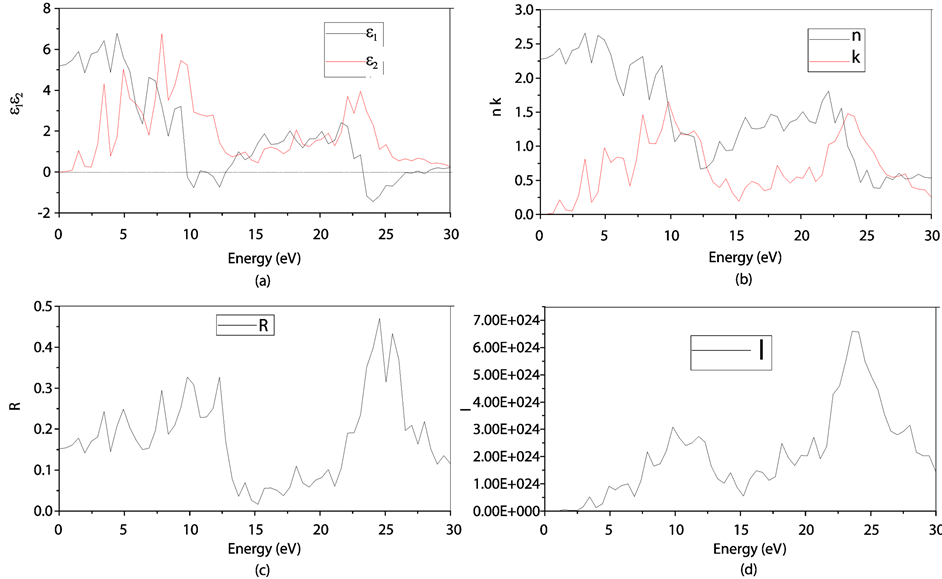
Figure 8. (a): The black and red lines represent our calculated real and imaginary parts of dielectric function ε(ω) respectively of O defect SrTiO2.875 (2 × 2 × 2) by LDA + U method; (b): The black and red lines represent our calculated real and imaginary parts of refractive index respectively of O defect SrTiO2.875 (2 × 2 × 2) by LDA + U method; (c): Optical spectra of the reflectivity R of O defect SrTiO2.875 (2 × 2 × 2) by LDA + U method; (d): Optical spectra of the adsorption coefficient I of O defect SrTiO2.875 (2 × 2 × 2) by LDA + U method
图8. LDA + U方法下SrTiO2.875 (2 × 2 × 2)的介电函数(a)、折射率(b)、反射率(c)、吸收系数(d)

Figure 9. Optical spectra of the adsorption coefficient I of O defect SrTiO2.875 (2 × 2 × 2) by LDA + U method
图9. LDA + U方法下SrTiO2.875 (2 × 2 × 2)放大后的吸收系数图
致谢
本课题由国家自然科学基金(10804095,61066005,11164032)和教育部新世纪优秀人才支持计划(NCET-12-1080)资助。感谢云南大学高性能计算中心的支持。
参考文献 (References)
- [1] T. Tanaka, K. Matsunaga, Y. Ikuhara, et al. (2003) First-principles study on structures and energetics of intrinsic vacancies in SrTiO3. Physical Review B, 68, Article ID: 205213.
- [2] Van Benthem, K., Elsasser, C., French, R. (2001) Bulk electronic structure of SrTiO3: Experiment and theory. Journal of Applied Physics, 90, 6156.
- [3] Kresse, G. and Furthmuler, J. (1996) Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Physical Review B, 54, 11169-11186.
- [4] Kresse, G., Furthmuler, J. (1996) Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Computational Materials Science, 6, 15-50.
- [5] Perdew, J.P., Burke, K. and Ernzerhof, M. (1996) Generalized Gradient Approximation Made Simple. Physical Review Letters, 77, 3865-3868.
- [6] Anisimov, V., Zaanen, J. and Andersen, O. (1991) Band Theory and Mott Insulators: Hubbard U instead of Stoner I. Physical Review B, 44, 943-954.
- [7] Dudarev, S.L., Botton, G.A., Savrasov, S.Y., et al. (1998) Electron-Energy-Loss Spectra and the Structural Stability of Nickel Oxide: An LSDA+U Study. Physical Review B, 57, 1505.
- [8] An, H.-S., Cuong, D.D., Lee, J.C. et al. (2006) LDA+U Study on Fully Relaxed LaTiO3 and (SrTiO3)m(LaTiO3)n Superlattice Structures. Journal of the Korean Physical Society, 49, 1536-1542.
- [9] Kowalczyk, S., McFeely, F., Ley, L., et al. (1977) The electronic structure of SrTiO3 and some simple related oxides (MgO, A12O3, SrO, TiO2). Solid State Communications, 23, 161-169.
- [10] Battye, F., Höchst, H. and Goldmann, A. (1976) Photoelectron studies of the BaTiO3 and SrTiO3 valence states. Solid State Communications, 19, 269-271.
- [11] Luo, W., Duan, W., Louie, S., et al. (2004) Structural and electronic properties of n-doped and p-doped SrTiO3. Physical Review B, 70, Article ID: 214109.
- [12] Guo, X., Chen, X., Sun, Y., et al. (2003) Electronic band structure of Nb doped SrTiO3 from first principles calculation. Physics Letters A, 317, 501-506.
- [13] 徐新发, 邵晓红 (2009) Y掺杂SrTiO:晶体材料的电子结构计算. 物理学报, 3, 1908-1916.
- [14] 赵庆勋, 温梦仙, 王书彪, 等 (2009) BiFeO3及Bi2FeCrO6的电子结构和光学特性. 人工晶体学报, 37, 6.
- [15] 李沛娟, 周薇薇, 唐元昊, 等 (2010) CeO2的电子结构, 光学和晶格动力学性质第一性原理研究. 物理学报, 59, 5.
- [16] 王渊旭, 王春雷, 钟维烈, 等 (2003) SrHfO3和SrTiO3光学特性的第一性原理研究. 物理学报, 53, 1000-3290.
- [17] Zhong, Z.C. and Kelly, P.J. (2008) Electronic-structure induced reconstruction and magnetic ordering at the LaAlO3/SrTiO3 interface. EPL, 84, 27001.

NOTES
*通讯作者。